
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Electronic properties and topological phases of ThXY (X = Pb, Au, Pt and Y = Sb, Bi, Sn) compounds
[Nourbakhsh Zahra, Vaez Aminollah†,  ]
]
]


† Corresponding author. E-mail:
The electronic properties and topological phases of ThXY (X = Pb, Au, Pt, Pd and Y = Sb, Bi, Sn) compounds in the presence of spin–orbit coupling, using density functional theory are investigated. The ThPtSn compound is stable in the ferromagnetic phase and the other ThXY compounds are stable in nonmagnetic phases. Band structures of these compounds in topological phases (insulator or metal) and normal phases within generalized gradient approximation (GGA) and Engel–Vosko generalized gradient approximation (GGA_EV) are compared. The ThPtSn, ThPtBi, ThPtSb, ThPdBi, and ThAuBi compounds have topological phases and the other ThXY compounds have normal phases. Band inversion strengths and topological phases of these compounds at different pressure are studied. It is seen that the band inversion strengths of these compounds are sensitive to pressure and for each compound a second-order polynomial fitted on the band inversion strengths–pressure curves.
ThXY (X = Pb, Au, Pt, Pd and Y = Sb, Bi, Sn) compounds have cubic MgAgAs crystal structure with 
There have been some empirical calculations of the structural and electronic properties of ThPtSn compound.[3] Szajek et al.[4] used the van Barth–Hedin exchange correlation potential and the tight-binding linear muffin–tin orbital method in the atomic sphere approximation to calculate the electronic density of states of ThPtSn compound and used the x-ray photoemission spectra to measure the lattice parameters and the valence band of this compound. They concluded that the ThPtSn compound has a small energy gap of 32 meV, which is sensitive to details of the method being used for the exchange–correlation potential. Grytowska et al.[5,6] measured the lattice parameters and magnetization isotherms of ThPtSn compound with magnetometer between 1.9 K and 300 K with the magnetic field strength up to 5 T. They did not observe any splitting for Sn and Pt, due to large Pt–Sn interatomic distance. Palstra et al.[7] used adiabatic heat pulse technique to measure the specific heat of ThPtSn compound and used x-ray diffraction to measure the lattice parameter and Th–Th distance of this compound. They used a Foner vibrating sample magnetometer operating to determine the magnetization of ThPtSn compound, used the adiabatic heat pulse technique to measure the specific heat of this compound, and found that ThPtSn exhibits a positive magnetoresistivity and obeys the quadratic field dependence between 4 K and 100 K. To our knowledge there are no experimental and theoretical works for other ThXY compounds.
Topological phases (insulators or metals) are novel states of quantum matter in which surface states are protected by time-reversal symmetry. These states are topologically protected against back scattering. These materials have very promising technological applications in quantum computing[8] and spintronics.[9,10]
The first goal of this paper is to compare and investigate the electronic properties of ThXY compounds within generalized gradient approximation (GGA)[11] and Engel–Vosko generalized gradient approximation (GGA_EV)[12] at zero pressure. Since some physical properties of ThPtSn compound at zero pressure have already been studied by others,[1–7] the ThPtSn calculated results are compared with the previous works and we have focused on the topological phase of this compound that has not been studied by others. In addition, the topological phases of other ThXY compounds are investigated. The second goal of this paper is to study the effect of pressure on the topological phases of ThXY compounds.
The results of this paper were attained based on the density functional theory (DFT) using the full potential linearized augmented plane wave plus local orbitals (LAPW+lo) method as implemented in the Wien2k package.[13] In LAPW+lo method, every unit cell is portioned into non-overlapping muffin-tin (MT) spheres with radii RMT and interstitial region. The muffin-tin radii of Th, X, and Y atoms were selected as 2.4 a.u., 2.3 a.u., and 2.3 a.u., respectively. The Kohn–Sham (KS) wave functions, charge density, and potential are expanded with spherical harmonics within the MT spheres and also with plane waves inside the interstitial region. In this paper, the maximum quantum number for atomic wave functions within the MT sphere was selected as lmax = 10. The wave vector cut-off for the plane wave expansion within the interstitial region was Kmax = 8/RMT (a.u.)−1. The charge density as well as the potential was Fourier expanded inside the interstitial region up to Gmax = 15 (Ry)1/2. 165 k-points were generated within the irreducible wedge of the Brillouin zone. The values of the lmax, Kmax, Gmax and the number of k-points were chosen through converging the total energy.
The exchange correlation potential was treated within GGA using the scheme of Perdew–Burke–Ernzerhof[11] and GGA_EV[12] approaches. The GGA_EV approach is based on potential optimized that has been developed for calculation of the energy band structure. The computation for the core electrons was carried out full relativistically, while for the valence electrons it was done both scalar and completely relativistically. The spin orbit coupling for the valence electrons was incorporated as a second-variational treatment[14] utilizing scalar relativistic eigenfunction as the basis.
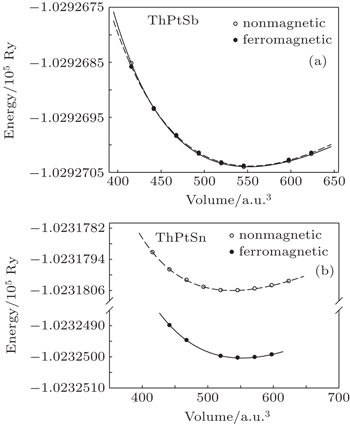
To investigate the ground state properties of ThXY (with X = Pb, Au, Pt, Pd and Y = Sb, Bi, Sn) compounds, the total energy as a function of the unit cell volume in ferromagnetic and nonmagnetic phases of these compounds is calculated using GGA. The calculated total energy–volume is fitted with the Murnaghan equation of state.[15] Due to the close similarity between the total energy–volume curves behavior of these compounds, only the curves for ThPtSn and ThPtSb compounds are shown in Fig.
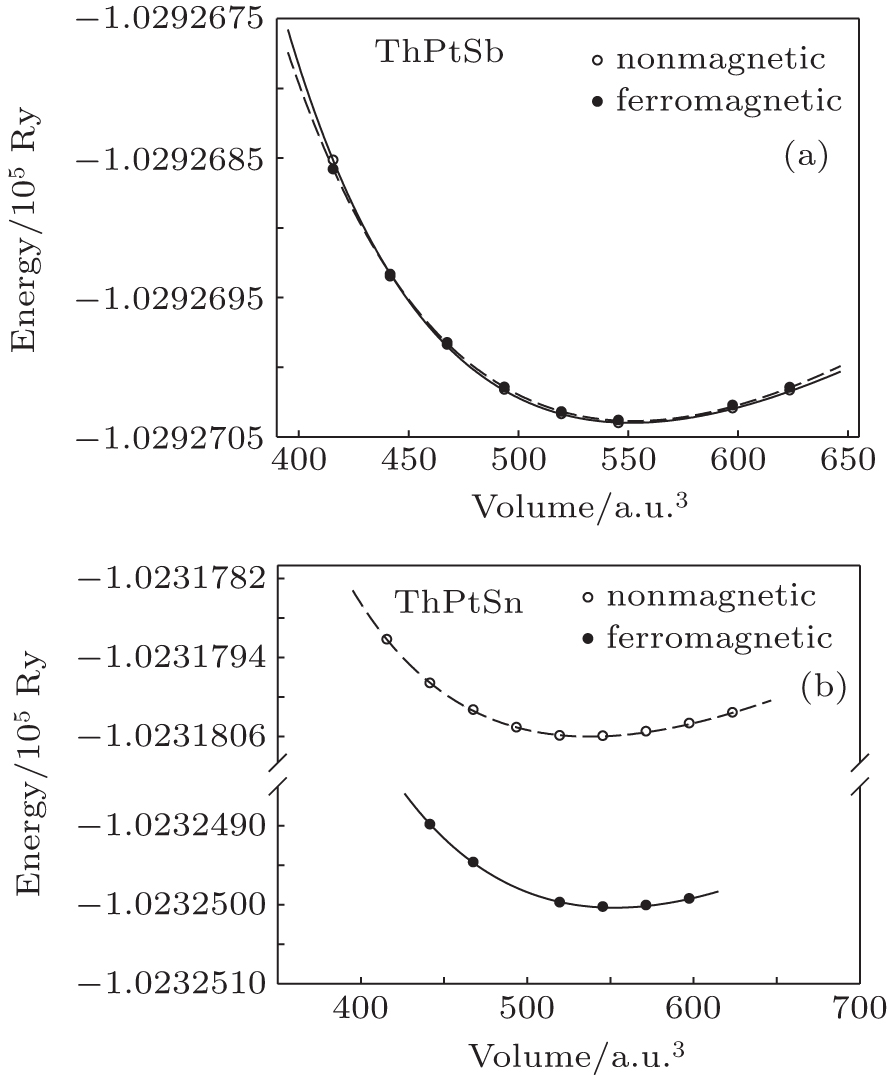 | Fig. 1. The calculated total energies of ThPtSn (a) and ThPtSb (b) compounds as a function of unit cell volume in ferromagnetic and nonmagnetic phases. |
| Table 1. The calculated lattice parameters (in Å) and the bulk moduli (in GPa) of ThXY compounds. . |
The equilibrium unit cell volume of matter is related to the size of the component atoms with some adjustment for bonding in the solid. Going upward in a column of the periodic table, the bonding contribution decreases by a small value. The calculated lattice parameters of ThPdY (Y = Sb, Bi, Sn) compounds are larger than the corresponding values of ThPtY compounds. For a given row in the periodic table, the lattice parameter should increase with valence number increasing. The calculated lattice constants of ThXY compounds show that the lattice constants of ThXSb are a little larger than the corresponding values of ThXSn. This is due to the increase of the number of valence electrons, when Sn atom in ThXSn compounds is replaced with Sb. Furthermore, we also calculated the bulk moduli of these compounds. Al-Douri et al.[16–18] and Nourbakhsh[19] have shown that the dominant effect on the bulk moduli is from the degree of covalency and ionicity. The bulk modulus generally increases with the increase of covalency and the decrease of ionicity. The effect of decreasing ionicity is to increase the amount of bonding charge and hence to increase the bulk modulus. ThXY compounds have different degrees of covalent and ionic bonding. The calculated bulk moduli of these compounds show as follows.
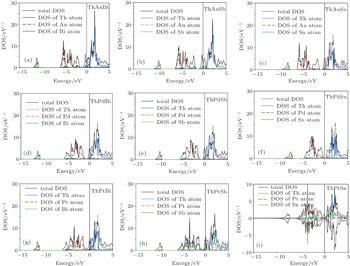
To study the electronic properties of ThXY compounds, we have calculated the total electron density of states (DOS) within GGA and GGA_EV approaches. The calculated results indicate that the spin–orbit interaction has a considerable effect and cannot be ignored. Thus, we have studied the electronic properties and topological phases of these compounds in the presence of spin–orbit coupling. Due to close similarity between the calculated results within GGA and GGA_EV, the results within GGA are only shown in Fig.
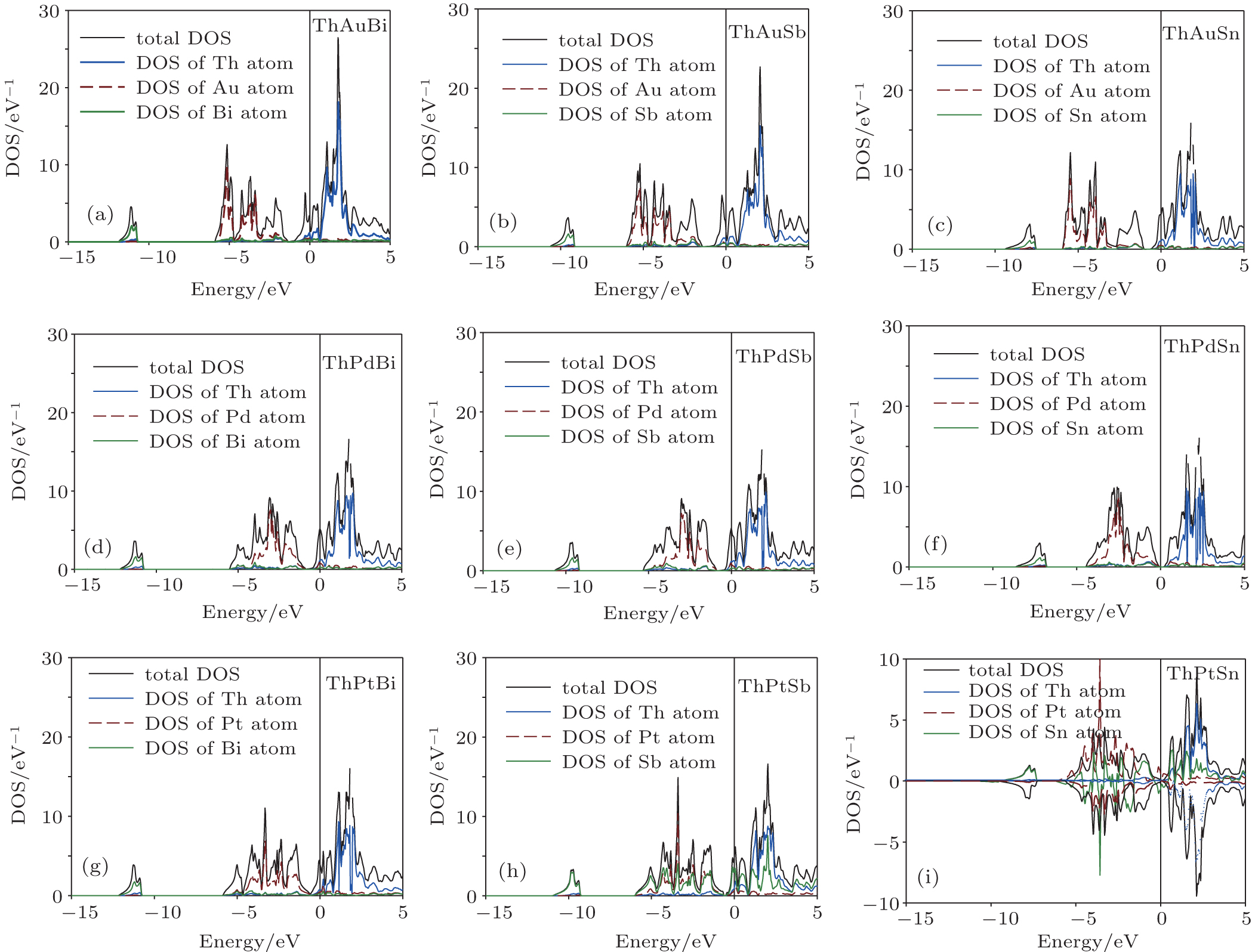 | Fig. 2. The calculated total and partial electron densities of states of ThXY compounds within GGA approach. (a) ThAuBi, (b) ThAuSb, (c) ThAuSn, (d) ThPdBi, (e) ThPdSb, (f) ThPdSn, (g) ThPtBi, (h) ThPtSb, (i) ThPtSn. |
The band structures of these compounds within GGA approach are calculated. The calculated results show that in ThPtSn and ThPdSn, the valence band’s maximum and the conduction band’s minimum occur at the Γ point; hence these two semiconductor compounds have small direct energy band gaps. The calculated energy band gap and available experimental value are given in Table
| Table 2. The calculated energy band gaps (eV) of ThPtSn and ThPdSn compounds within GGA and GGA_EV and experimental results. . |
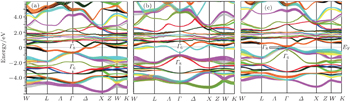
In band structure calculation, based on the DFT, the GGA usually underestimates the energy band gap. Engel and Vosko,[12] in the GGA_EV improved the GGA for band structure calculations, with optimizing exchange potential (Vx). In some previous papers[19–21] the GGA_EV was applied for band structure calculation of solids and compared the results with GGA calculations. It has been shown that the GGA_EV improves the determination of band gap and some other electronic properties. We have calculated the band structure of ThXY compounds within GGA_EV using the calculated equilibrium lattice parameters (as obtained in Section 3.1) within GGA in the presence of spin–orbit coupling. Due to the close similarity between the band structures of these compounds, the band structures are only given for ThPtBi, ThPdSb, and ThPtSn compounds in Fig.
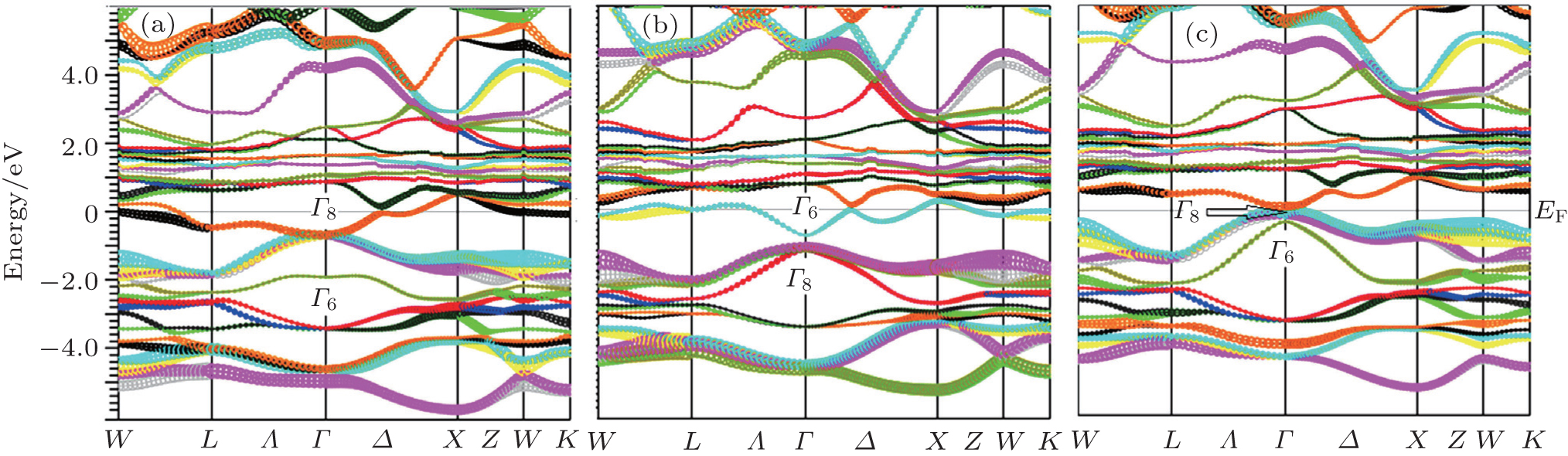 | Fig. 3. The calculated band structures of ThPtBi (a), ThPdSb (b), and ThPtSn (c) compounds within GGA_EV approach. |
Using the calculated band structure of ThXY compounds in the presence of spin–orbit coupling, we have studied the band order of these compounds. The energy bands of these compounds at the Γ point near the Fermi energy split, due to the spin–orbit coupling, into Γ6 (s-like levels), Γ7 (p-like levels, j = 1/2) with twofold degeneracies and Γ8 (p-like levels, j = 3/2) with fourfold degeneracy. The band orders of ThPtSb and ThPdSb are from high to low energy Γ8, Γ6, Γ7 and Γ6, Γ8, Γ7, respectively. The ThPdSb has a normal band order (the Γ6 state sits above the state), while the ThPtSb has an inverted band order (the Γ6 state sits below the Γ8 state).
The topological phases of these compounds can be characterized by band inversion between Γ6 and Γ8 at the Γ symmetry point in the first Brillouin zone. The band inversion strength (Δ) in compounds with cubic symmetry is defined as the energy difference between Γ6 and Γ8 states (Δ = EΓ6 − EΓ8). A negative Δ indicates that the compound is a topological nontrivial insulator, semimetal or metal, while that with a positive Δ is a topological trivial phase.
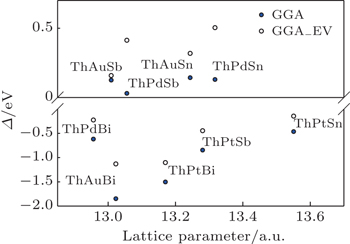
The band inversion strengths of ThXY compounds using the calculated band structures (Fig.
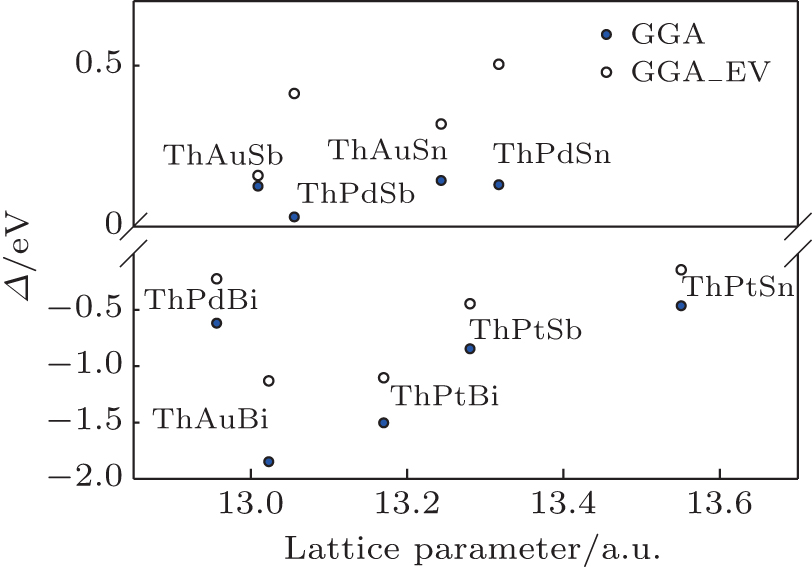 | Fig. 4. The calculated band inversion strengths, Δ, of ThXY compounds within GGA and GGA_EV approaches. |
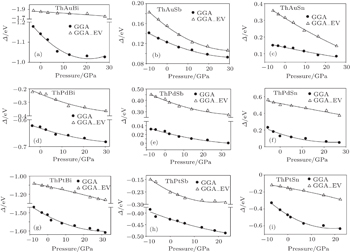
To study the effect of pressure on the band order of ThXY compounds, the band structures of these compounds within GGA and GGA_EV at different pressure are calculated. The GGA_EV approach is not a good approximation for energy–volume curve and pressure calculations,[19] so the pressure (the lattice parameter) was calculated within GGA and applied to the band structure calculations within GGA and GGA_EV approaches. The calculated Δ as a function of pressure within GGA and GGA_EV are shown in Fig.
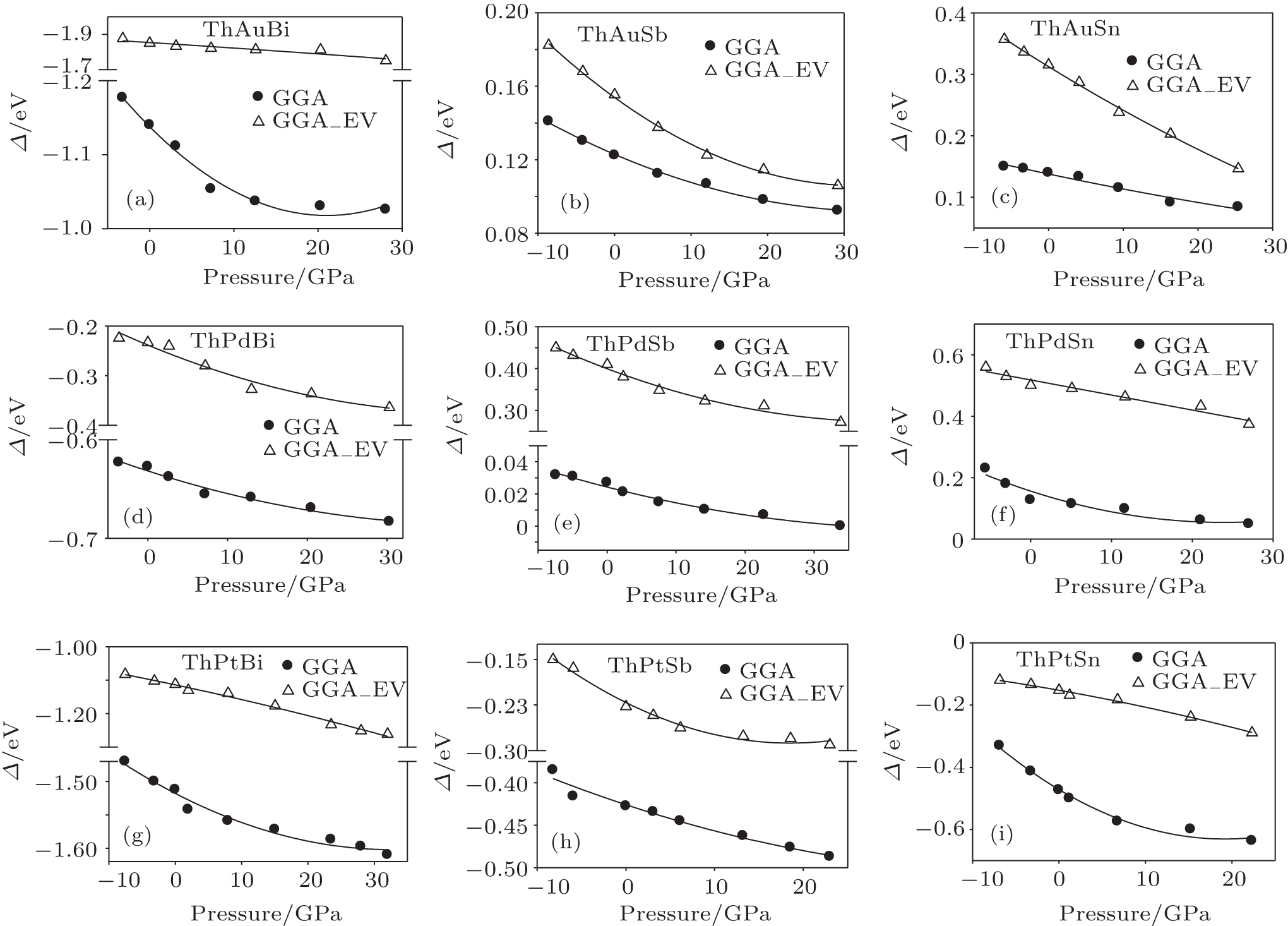 | Fig. 5. The calculated band inversion strength (Δ) as a function of pressure for different compounds within GGA and GGA_EV. (a) ThAuBi, (b) ThAuSb, (c) ThAuSn, (d) ThPdBi, (e) ThPdSb, (f) ThPdSn, (g) ThPtBi, (h) ThPtSb, (i) ThPtSn. |
| Table 3. The calculated first-order (α) and second-order (β) pressure coefficients of band inversion strength within GGA and GGA_EV.. . |
We have studied the band structures of ThXY compounds using GGA and GGA_EV approaches for the purpose of delineating their band order and topological phases. ThPtSn, ThPtSb, ThPtBi, ThPdBi, and ThAuBi compounds have an inverted band order, while ThPdSb, ThPdSn, ThAuSn, and ThAuSb compounds have a normal band order. ThPtSn is a candidate for the topological insulator and ThPtSb, ThPtBi, ThPdBi, and ThAuBi are candidates for topological metals.
The behavior of band inversion strength of these compounds with pressure shows that the variation of band inversion strength is not linear and changes with pressure by a second-order polynomial. The second-order pressure coefficients of these compounds are smaller than the first-order pressure coefficients.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 |