{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Energetics of carbon and nitrogen impurities and their interactions with vacancy in vanadium
[Hua Juan1, 2, Liu Yue-Lin2, Li Heng-Shuai1, 3, Zhao Ming-Wen1, Liu Xiang-Dong1, †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 11575153 and 11375108).
We studied the energetic behaviors of interstitial and substitution carbon (C)/nitrogen (N) impurities as well as their interactions with the vacancy in vanadium by first-principles simulations. Both C and N impurities prefer the octahedral site (O-site). N exhibits a lower formation energy than C. Due to the hybridization between vanadium-d and N/C-p, the N-p states are located at the energy from −6.00 eV to −5.00 eV, which is much deeper than that from −5.00 eV to –3.00 eV for the C-p states. Two impurities in bulk vanadium, C–C, C–N, and N–N can be paired up at the two neighboring O-sites along the 〈111〉 direction and the binding energies of the pairs are 0.227 eV, 0.162 eV, and 0.201 eV, respectively. Further, we find that both C and N do not prefer to stay at the vacancy center and its vicinity, but occupy the O-site off the vacancy in the interstitial lattice in vanadium. The possible physical mechanism is that C/N in the O-site tends to form a carbide/nitride-like structure with its neighboring vanadium atoms, leading to the formation of the strong C/N–vanadium bonding containing a covalent component.
In the fusion reactor, some components, such as the first wall, the divertor, and the structural materials exposed to plasma particles and electromagnetic radiation, will be irradiated by high energy neutrons,[1–6] which induces the degradation of the series properties and the residual radioactivity of the exposed materials. In order to efficiently utilize the fusion power, tremendous efforts have been devoted to searching for the suitable structural materials in the last few decades.[7–15]
In many experimental studies, vanadium and vanadium-based alloys have been attested to be promising structural materials owing to their excellent performances at high temperatures,[16–19] such as the intrinsic low-activation characteristics, high-temperature strength, and high thermal stress factor,[17] all of which can satisfy the requirements of safety operation in the extreme conditions. However, some small atomic-radius elements such as carbon (C) and nitrogen (N) are always present in vanadium-based materials, either as alloying elements in vanadium or as impurities in pure vanadium solid. We already knew that once beyond their solubility limit, C and N impurities tend to precipitate as carbides and nitrides, further leading to a notable change of the mechanical properties such as hardening and embrittlement. Moreover, even the high-purity vanadium can also contain a few atomic parts per million (appm) of these impurities. Several experimental and theoretical studies have demonstrated that C and N are the most frequent foreign interstitial atoms in the realistic vanadium and vanadium-based alloy materials.[20–26] Experimentally, some basic behaviors of C and N in vanadium-based alloys have been investigated. For example, in Ref. [22], Diercks et al. investigated the correlation between the mechanical properties of vanadium-based alloys and the impurity levels of C and N. Through the thermal desorption spectroscopy experiments,[23,24] it was found that C and N impurities in the HenVnX-type defects (X = C/N), namely, carbides and nitrides, could influence He dissociation from the HenVnX-type complexes remarkably. It was also pointed out in Ref. [25] that the reduction of C in the grain boundaries can affect the ductility of vanadium alloys. Recently, Li et al. investigated the stability and diffusion behaviors of a C or N impurity in vanadium solid using first-principles simulation method.[26]
Although many studies have investigated the behaviors of C or N impurity in vanadium or vanadium alloys as mentioned above, to the best of our knowledge, the C–N interaction and the interplay of C or N impurity atoms with the vacancy in vanadium have not yet been explored on the theoretical side till now, especially the binding properties between C or N impurity and vacancy. Several experiments have shown that at concentrations as low as a few appm, C and N might already have significant influences on the structural defects, such as vacancies in other metals.[27–30] The observed effects generally originate from an attractive interaction between the impurity atoms and the vacancies. Even though the solubility limits are low, the interactions of C/N impurities with the vacancies are strong enough according to the previous theoretical predictions.[31–33] Motivated by this, here we employ first-principles simulations to investigate the C–N interaction and their interactions with a single vacancy in the vanadium solid in detail. Our results indicate that, differing from the previous results,[31–33] both C and N impurities in vanadium do not like to stay at the vacancy vicinity but at an octahedral interstitial site outside of the vacancy. This kind of interesting phenomenon mainly stems from that C and N impurities prefer to form the covalent-like bonding with the two first nearest-neighbor (1nn) vanadium atoms.
The first-principles calculations were performed using the Vienna ab-initio simulation package (VASP) code,[34,35] which is based on the density functional theory. The Perdew and Wang (PW91) functional[36] within the generalized gradient approximation for the exchange–correlation interaction and the projector augmented wave potentials[37,38] were adopted. In the current work, a supercell composed of 128 atoms (4×4×4) was used to model the vanadium solid, and the plane wave energy cutoff was set to be 350 eV, which is sufficient for the calculations of the total energy and the geometry of the supercell. The Brillouin zone was sampled with a 3×3×3 k-point mesh generated by the Monkhorst–Pack scheme.[39] All of the atomic coordinates and volumes were relaxed with the conjugate gradient method until the forces on all the atoms in our calculations were less than 10−3 eV ·Å−1. The Methfessel–Paxton smearing method[40] was employed to integrate the Brillouin zone and account for the partial occupancy of the metals near the Fermi level with a smearing width of 0.1 eV, which keeps the convergence error of the total energy below 1 MeV. The equilibrium lattice constant of the bcc vanadium obtained from the present calculations is 2.98 Å, which is in excellent agreement with various experimental data ranging from 2.99 Å to 3.00 Å.[41–44]
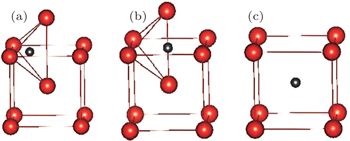
In bcc metal, there are two kinds of typical point defects, namely, the substitution defects and the interstitial ones. For simplicity, we here only consider two typical high symmetrical interstitial sites, i.e., tetrahedral and octahedral sites, and set all substitution sites to be equivalent, as illustrated in Fig.

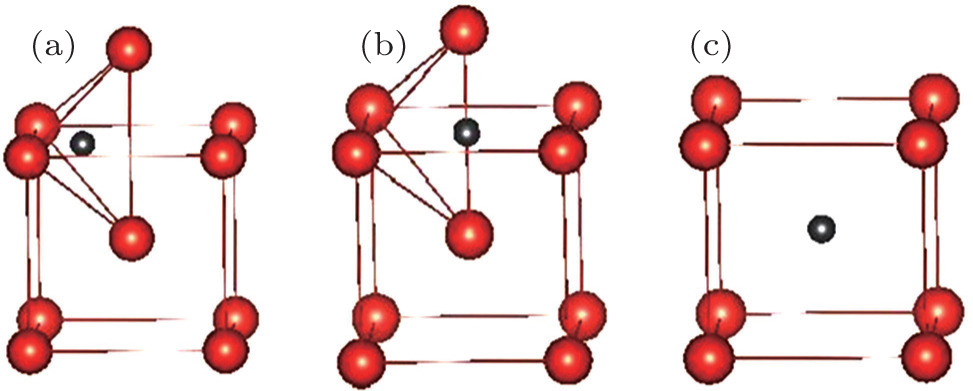 | Fig. 1. Three possible positions for an impurity atom in vanadium: (a) tetrahedral site (T-site), (b) octahedral site (O-site), and (c) substitution site (S-site). Small (gray) and large (red) balls represent impurity atom (C or N) and vanadium atom, respectively. |
In Table
| Table 1. Formation energy (in eV) of impurity (C or N) at different possible position in bulk vanadium. . |
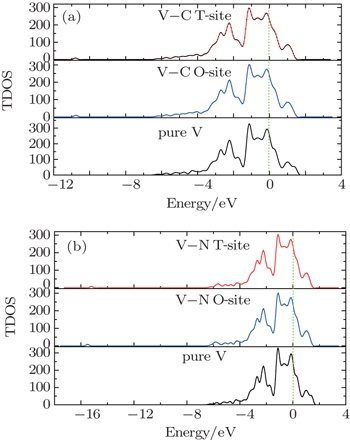
In Fig.
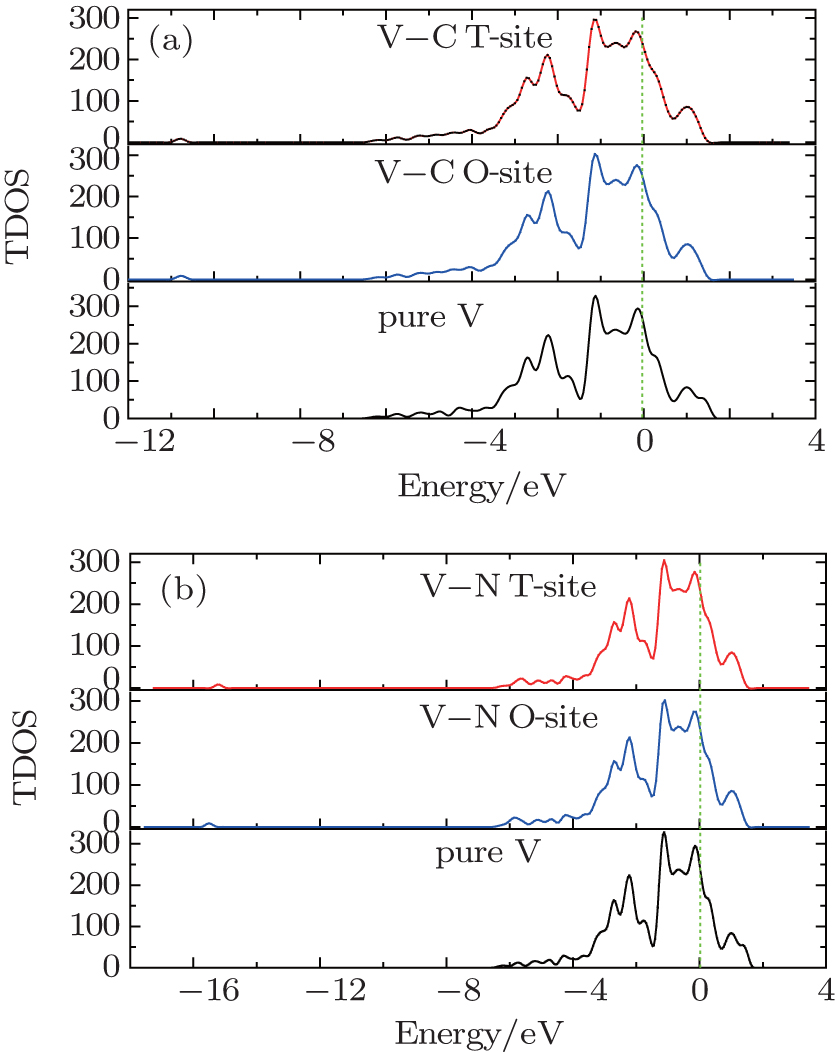 | Fig. 2. The total DOS of vanadium without and with the impurity atom at the T-site and the O-site: (a) impurity C case, (b) impurity N case. The Fermi energy is set to be zero in all cases. |
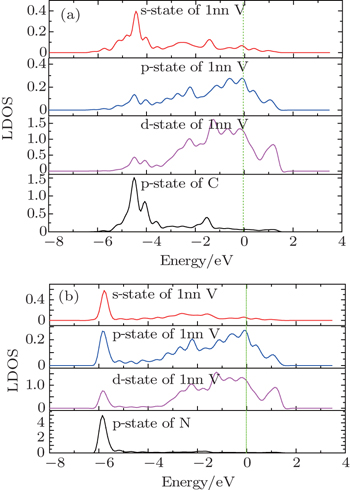
In order to attest the formation of covalent bonding between C/N and vanadium atoms, we further calculated the local density of states (LDOS) of C/N for impurity C/N at the O-site because the O-site is lower in formation energy of C/N than the T-site. As seen from Fig.
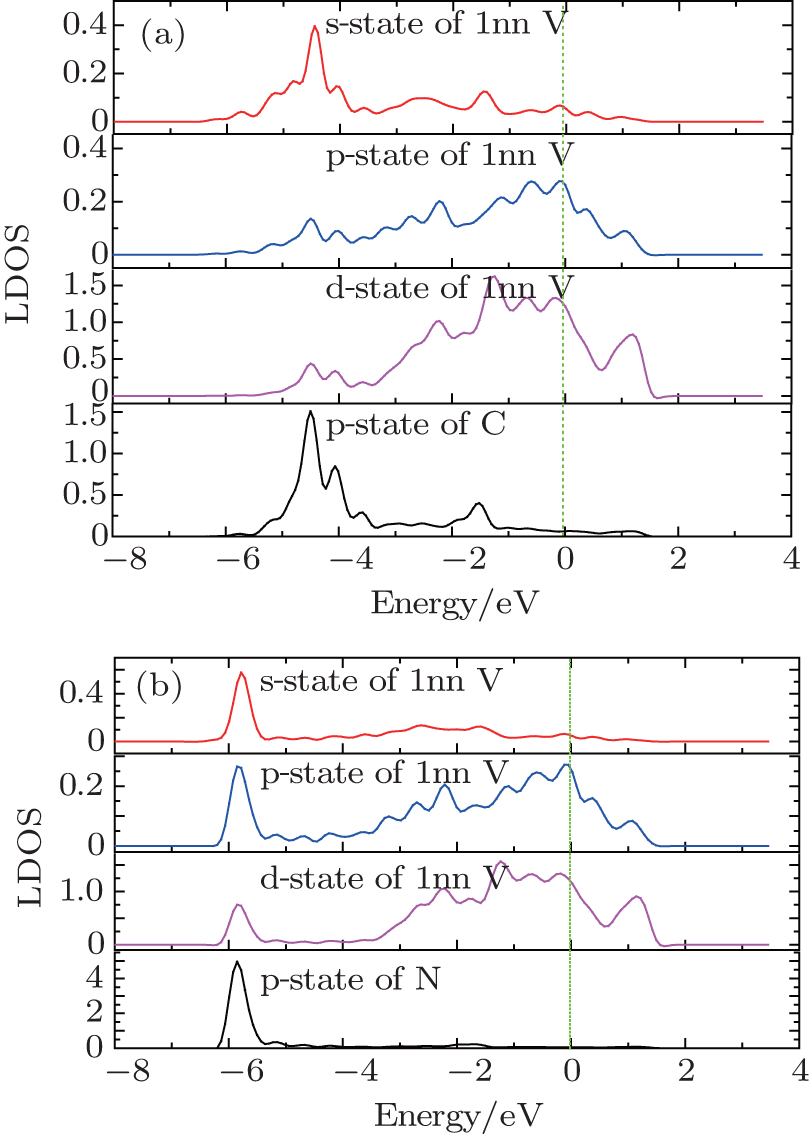 | Fig. 3. The LDOS of 1nn vanadium atom of impurity C/N atom after the C/N occupying the O-site in vanadium solid: (a) impurity C case, (b) impurity N case. |
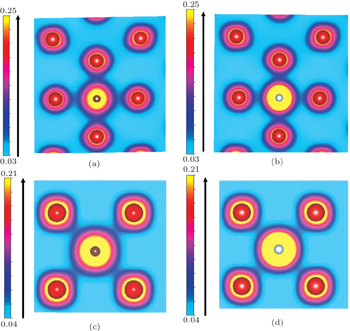
To obtain further insight into the interaction between an interstitial impurity and its neighboring vanadium atoms, we display the charge density distribution for an individual C or N atom placed in the O-site of the vanadium solid in Fig.
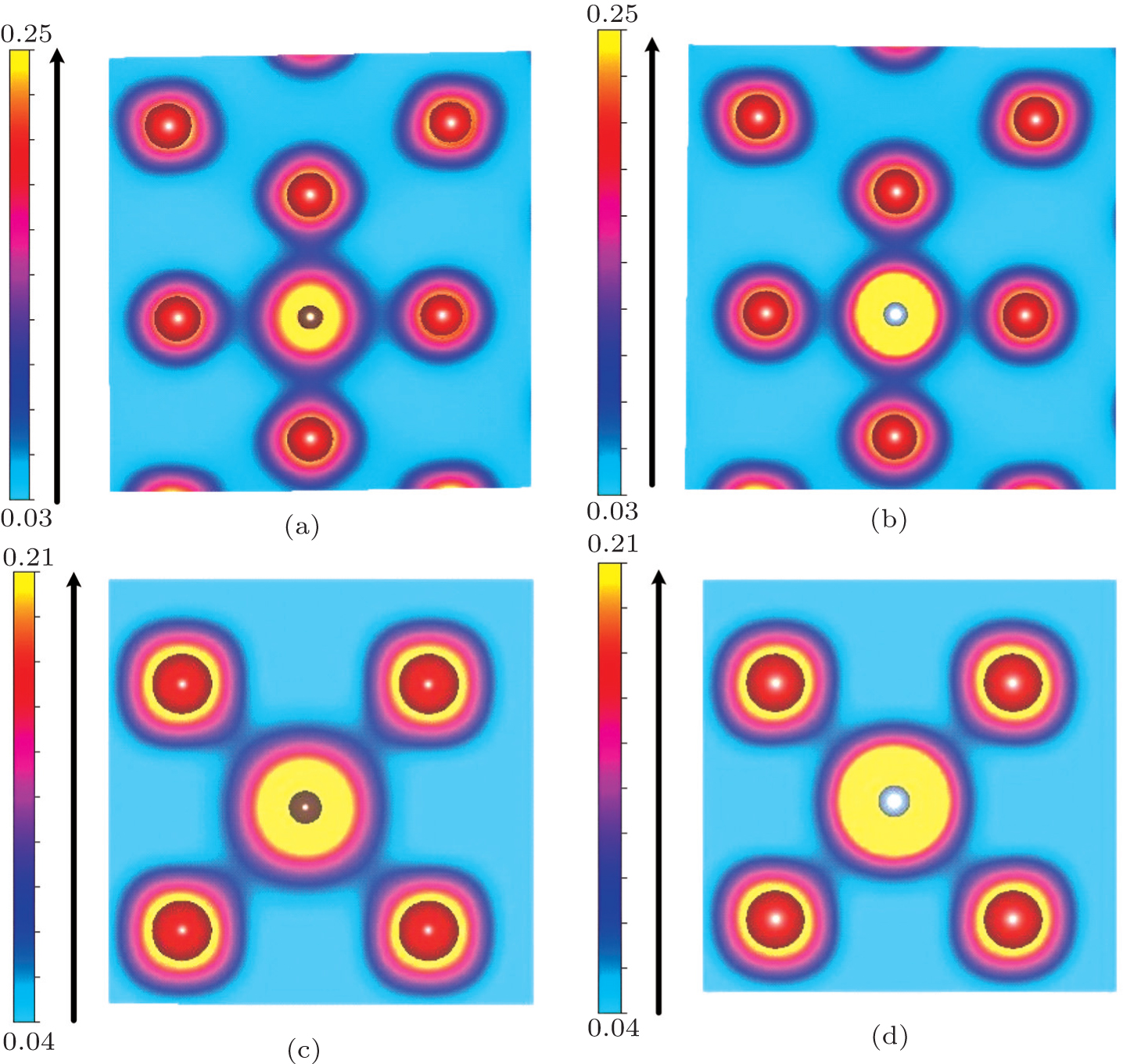 | Fig. 4. The charge density distributions of impurity at an octahedral position in the (010) plane: (a) C with 1nn vanadium atoms, (b) N with 1nn vanadium atoms, (c) C with 2nn vanadium atoms, (d) N with 2nn vanadium atoms. |
Our theoretical results show that the O-site is the most stable position for single C or N atom in bcc vanadium solid. As is well known, in a real material, there is a high possibility for the co-existence of impurities in the nearby interstitial positions. Thus, we turn to address the impurity–impurity interactions. According to the impurity formation energies in vanadium (Table
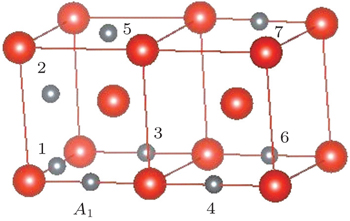
We construct seven possible configurations containing two impurities including C–C, C–N, and N–N pairs in bcc vanadium. As presented in Fig.
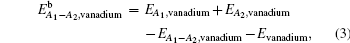
 | Fig. 5. Seven possible configurations for two impurities in bcc vanadium. A1 is one O-site occupied by the first impurity. The second impurity atom will be located at different O-sites labeled by i (i = 1, 2, …, 7). |
| Table 2. Binding energies (in eV) of A1–A2 pairs in bulk vanadium with different configurations as shown in Fig. |
We further observe that the C–C pair is significantly less repulsive than the N–N pair at the shorter distances, i.e., from cfg1 to cfg4. At the larger distance, i.e., from cfg5 to cfg7, the C–C interaction is either less or more repulsive than the N–N pair. It is also worth mentioning that the C–C pair can also be formed when two C atoms are located at the two respective O-sites along the 〈010〉 direction with the distance of 1.000 Å (cfg3 in Fig.
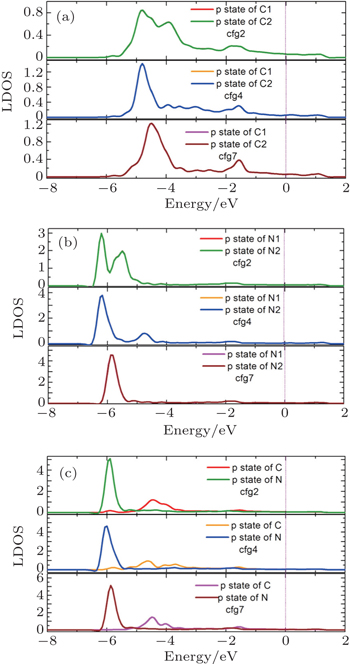
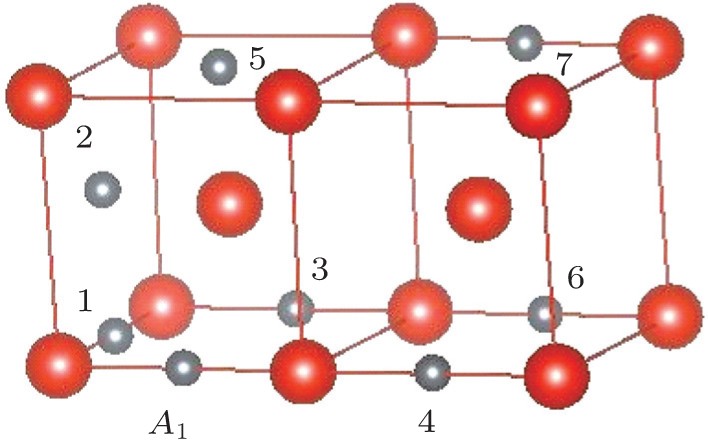
In order to gain further insight into the stability of C–C, N–N, and C–N pairs, we then compare the LDOSs of these pairs with different atom–atom distances in vanadium. As shown in Figs.
 | Fig. 6. Comparison of LDOSs of the C–C, N–N, and C–N pairs in the cfg2, cfg4, and cfg7 configurations (see these configurations in Fig. |
Generally, when an impurity is dissolved in a solid metal, it can be easily captured by vacancies. Further, with the increase of impurity atoms, they tend to accumulate to form the impurity–vacancy clusters. According to the current calculations, however, we discover that both C and N impurities do not prefer to stay at the vacancy center and its vicinity, but occupy the O-site that is out of the vacancy. Such anomalous behavior is because the C/N impurity occupies the O-site outside the vacancy to form a vanadium–carbide/vanadium–nitride structure. Below, we will present the detailed discussion.
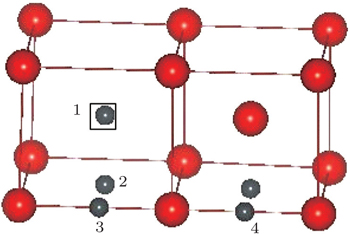
Figure 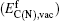
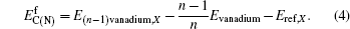
 | Fig. 7. Four possible sites for one C/N atom in the vicinity of one vacancy in vanadium. The larger red balls, the smaller gray balls, and the open square represent vanadium atoms, C/N atoms, and vacancy center, respectively. |
| Table 3. Formation energy (in eV) of one impurity (C/N) atom around a vacancy in vanadium with a different configuration given in Fig. |
Table
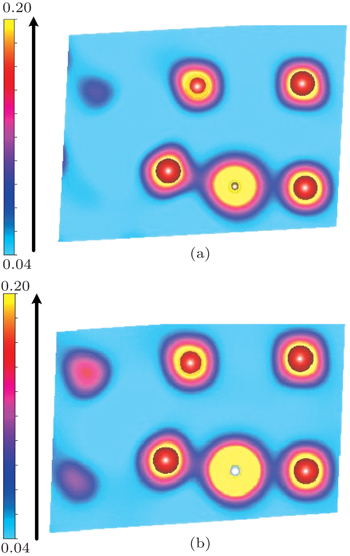
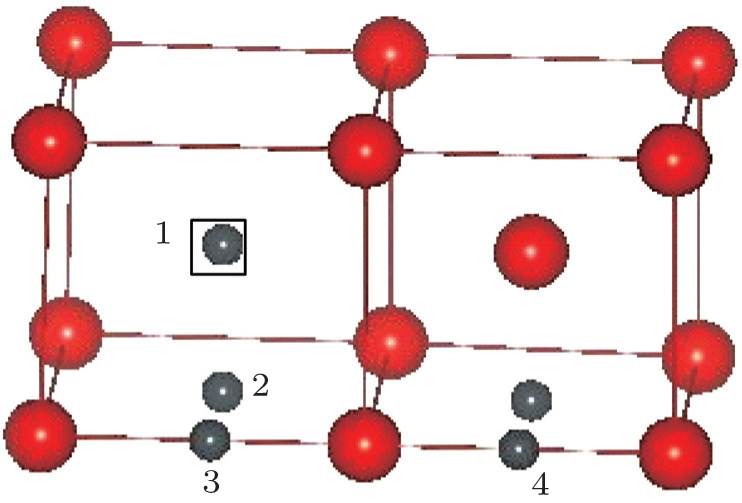
Now, we discuss the possible physical origin underlying the behavior of the C/N–vacancy interaction in bcc vanadium. It is important to realize that, according to the normal understanding, a vacancy in most metals can open up more space (i.e., free volume) to bind any given solute such as hydrogen,[54–56] helium,[57–59] oxygen,[60–62] carbon,[63] and so on. Whereas in vanadium, the energy minimization finds the most stable position for the C/N solute to be at an O-site, which is off vacancy-center/vicinity (site 4 presented in Fig.
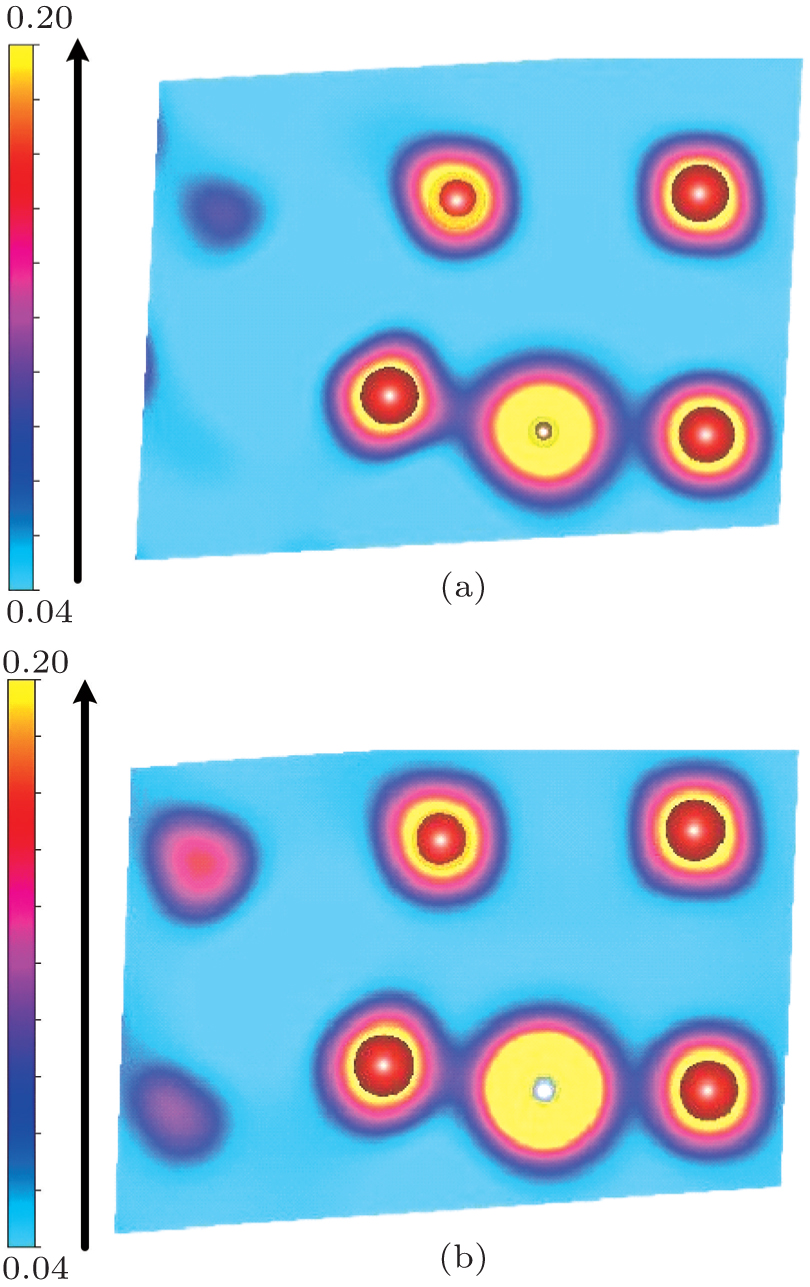 | Fig. 8. The charge density distributions in the (110) plane for (a) C and (b) N at the O-site, which is out of the vacancy. The larger red balls, the middle gray ball, and the smaller white ball denote vanadium atoms, C atom, and N atom, respectively. |
We have investigated the atomic geometry, formation energies, and electronic structure of interstitial and substitution carbon (C)/nitrogen (N) impurities as well as interactions of the two impurities with the vacancy in vanadium solid, employing the first-principles calculations. The results show that the octahedral site (O-site) for both C and N impurities is the lowest energy geometry structure in vanadium. In comparison with C, N exhibits the lower formation energy. Due to the hybridization between vanadium-d and N/C-p, the N-p states are located at the energy from −6.00 eV to −5.00 eV, which is much deeper than that from −5.00 eV to –3.00 eV for the C-p states. Regarding the interaction between two impurities in bulk vanadium, C–C, C–N, and N–N can be paired up at the two neighboring O-sites along the 〈111〉 direction and the binding energies of pairs are 0.227 eV, 0.162 eV, and 0.201 eV, respectively. Further, we find that both C and N do not prefer to stay at the vacancy center and its vicinity, but occupy the O-site off the vacancy in the interstitial lattice in vanadium. The possible physical mechanism is that C/N in the O-site tends to form a carbide/nitride-like structure with its neighboring vanadium atoms, leading to the formation of the strong C/N–vanadium bonding containing covalent component.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 | |
| 52 | |
| 53 | |
| 54 | |
| 55 | |
| 56 | |
| 57 | |
| 58 | |
| 59 | |
| 60 | |
| 61 | |
| 62 | |
| 63 | |
| 64 | |
| 65 | |
| 66 | |
| 67 | |
| 68 | |
| 69 |