Electronegativity explanation on the efficiency-enhancing mechanism of the hybrid inorganic–organic perovskite ABX 3 from first-principles study
[Chen Qing-Yuan1, Huang Yang1, Huang Peng-Ru1, Ma Tai1, Cao Chao2, He Yao1, †,  ]
]
]
 |


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 61366007, 11164032, and 61066005), the Program for New Century Excellent Talents in University of Ministry of Education of China (Grant No. NCET-12-1080), the Basic Applied Research Foundation of Yunnan Province, China (Grant Nos. 2011CI003 and 2013FB007), and the Excellent Young Talents in Yunnan University, China.
Organic–inorganic hybrid perovskites play an important role in improving the efficiency of solid-state dye-sensitized solar cells. In this paper, we systematically explore the efficiency-enhancing mechanism of ABX3 (A = CH3NH3; B = Sn, Pb; X = Cl, Br, I) and provide the best absorber among ABX3 when the organic framework A is CH3NH3 by first-principles calculations. The results reveal that the valence band maximum (VBM) of the ABX3 is mainly composed of anion X p states and that conduction band minimum (CBM) of the ABX3 is primarily composed of cation B p states. The bandgap of the ABX3 decreases and the absorptive capacities of different wavelengths of light expand when reducing the size of the organic framework A, changing the B atom from Pb to Sn, and changing the X atom from Cl to Br to I. Finally, based on our calculations, it is discovered that CH3NH3SnI3 has the best optical properties and its light-adsorption range is the widest among all the ABX3 compounds when A is CH3NH3. All these results indicate that the electronegativity difference between X and B plays a fundamental role in changing the energy gap and optical properties among ABX3 compounds when A remains the same and that CH3NH3SnI3 is a promising perovskite absorber in the high efficiency solar batteries among all the CH3NH3BX3 compounds.
The hybrid inorganic–organic perovskite absorbers ABX3 (A = CH3NH3; B = Sn, Pb; X = Cl, Br, I) have attracted lots of attention due to their outstanding performances as dye-sensitized solar-cell absorbers, topological insulators, and superconductors.[1–9] The researches of one type of the solar-cell absorber, the hybrid perovskite solar battery, have made great process in the last 5 years.[1–4] Solid-state dye-sensitized solar cells are composed of the semiconducting layer, the sensitizer, the transparent conducting and counter conducting electrodes, and the electrolyte. The working principle of solid-state dye-sensitized solar cells can be summed up as follows:
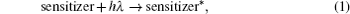
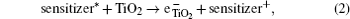
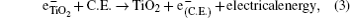
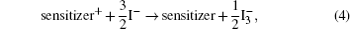
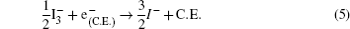
Specifically, we can explain these equations as follows. In order to provide the surface area to adsorb sensitizers, TiO2 is placed on the conducting electrode. When the photons irradiate on them, sensitizers are excited (Eq. (
The hybrid perovskite solar battery stands out due to its low cost, high efficiency, and environmental friendliness. Meanwhile, it is becoming a most promising battery compared with various solar batteries. Organic–inorganic hybrid CH3NH3PbX3 (X = Br, I) perovskites were introduced by Kojima et al. in 2009 as a new type of DSC sensitizing material.[11] Lee et al. reported a solution-processable solar cell with a conversion efficiency of 10.9%.[1] The solar conversion efficiency did not increase to 15% till recently, which was reported in a Nature paper by Burschka et al., and its high stability is equal to or even greater than those of the best thin-film photovoltaic devices.[2] As is well known, the ABX3 absorbers with high absorption coefficients will be excellent materials for enhancing the solar-converting efficiency of the solar battery. Previously, the properties of ABX3 were mostly investigated by several experimental studies which claimed that ABX3 is a great solar-cell absorber due to its symmetric structure and electronic properties.[7–13] Nevertheless, theoretical researches of ABX3 are rather rare and incomplete.[11–13]
In this study, we explore the efficiency-enhancing mechanism of ABX3 and systematically investigate the ABX3-type compounds which can stimulate the optical properties for hybrid inorganic–organic perovskite absorbers by first-principles calculations.[14,15] We find that the VBM and the CBM of the ABX3 are mainly composed of anion X p states and cation B p states respectively. The band gap of the ABX3 decreases and the absorptive capacities to different wavelengths of light expands when changing the B atom from Pb to Sn and changing the X atom from Cl to Br to I. Moreover, the organic framework A also plays an important role in ABX3. For instance, with the size of the organic framework A increasing, the band gap increases but atoms B and X remain the same. The changing trend of the band gap will be the same when the atoms B and X change and the size of framework A remains the same. So we just examine the situation where the organic framework A remains unchanged to investigate the efficiency-enhancing mechanism of ABX3 in this work. Finally, it is discovered that CH3NH3SnI3 has the best optical properties, and its light-adsorption range is the widest among all the ABX3 compounds when A remains the radical 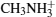
Our calculations were performed using the Vienna ab initio simulation package (VASP) with the projector-augmented wave (PAW) pseudopotentials.[16,17] We used the Perdew–Burke–Ernzerhof generalized gradient approximation to treat the exchange–correlation energy of interacting electrons.[18–20] The 2s, 2p electrons of C, 2s, 2p electrons of N, 1s electrons of H, 5d, 6s, 6p electrons of Pb, 2s, 2p electrons of Sn, and the 5s, 5p electrons of I were explicitly treated as valence electrons.[22,23] In order to guarantee the accuracy of the calculation, the energy cutoff for plane-wave cut-off is 500 eV. We used an A (6× 6× 6) Monkhorst–Pack k-point grid for geometry optimization and an A (10× 10× 10) grid for calculating the density of states. For the best approach to this calculation, the generalized gradient approximation (GGA) approach which is accurate and the time saving was adopted instead of the local density approximation (LDA) and HSE approach. All considered structures were fully optimized with the force on each atom being less than 0.05 eV/Å.[21,22]
ABX3 type compound shows different phases under different temperatures. In the high temperature phase, the crystal structure of all ABX3 adopts a cubic perovskite structure as shown in Fig. 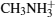
Structure of ABX3. X is shown in the vertex of the octahedron, B is shown in the center of the octabedron, the compound in the center is CH3NH3+.
The computations of optical properties were carried out by the perturbation theory method that was implemented in the VASP code. The main optical spectra, such as the refractive index N(ω), reflectivity R(ω), and adsorption coefficient I(ω), all can be obtained from the dynamical dielectric response functions (ɛ). The explicit expressions are given by
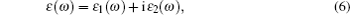
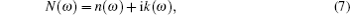
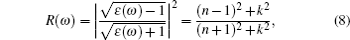
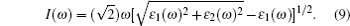
ABX3 compounds play a pivotal role in the perovskite sensitized solar cell mainly due to their optical properties which can induce the perovskite sensitized solar cells to absorb a wide range of light. According to Eqs. (
To find the material with the widest absorption range, the absorption coefficients of different ABX3 compounds were calculated. We tried both the GGA approach and the LDA approach, and their results are shown in Figs.
Optical spectra of the absorption coefficient of ABX3 (A = CH3NH3; B = Sn, Pb; X = Cl, Br, I), obtained by the GGA method.
Optical spectra of the absorption coefficient of ABX3 (A = CH3NH3; B = Sn, Pb; X = Cl, Br, I) by the LDA method.
Optical spectra of CH3NH3SnI3. In panel (a), the black and red curves represent real and imaginary parts of dielectric function ɛ (ω), respectively. In panel (b) the black and red curves represent real and imaginary parts of refractive index, respectively. Panel (c) shows the optical spectrum of the reflectivity R. Panel (d) displays the optical spectrum of the absorption coefficient.
Figure
To understand the trend of absorption edges of various ABX3 compounds, their electronic properties must be addressed. According to the experimental data which we could find, the GGA method is more accurate than the LDA as shown in Fig.
According to the analysis of the density of states (DOS), electronic states near the band gap are governed by the orbital overlap of the BX6 octahedron. In detail, as shown in Fig.
Figure
The band gaps and hence the optical edges of ABX3 compounds are governed by the electronegativity difference between the cation B and anion X. As the VBM is dominated by anion X components and the CBM is dominated by cation B, the separation between VBM and CBM is governed by the orbital interaction of the components. From a molecular theory point of view, the orbital interaction is closely related to the relative electronegativity between cation and anion. From this consideration, the results of the band gap shown in Fig.
The electronegativity difference between cation and anion also determines the bonding nature of the BX3 framework. As shown by the electron localization function (ELF) plotted in Fig.
Comparison of the band gaps of CH3NH3BX3 (B = Pb,Sn; X = Cl, Br, I), obtained by the LDA method (black line), and the GGA method (red line) with experimental results (blue line).
Density states of CH3NH3BX3 (B = Pb, Sn; X = Cl, Br, I).
Bandgaps corresponding to different matters of the ABX3.
The research on the relationship between the electronegativity and the optical and electronic properties of hybrid organic–inorganic perovskites could not only reveal the change regulation of ABX3 when the organic framework A is CH3NH3, but also provide a new way of explaining the basic physical efficiency-enhancing mechanism of all hybrid organic–inorganic perovskite structures ABX3.
Electronegativity by Pauling scale.
| Element | Electronegativity |
|---|---|
| Sn | 1.96 |
| Pb | 1.8 |
| Cl | 3.16 |
| Br | 2.96 |
| I | 2.66 |
ELF map of CH3NH3BX3 compounds.
In this work, we systematically investigate the optical and electronic properties of ABX3 compounds through density functional calculations. For the ABX3 compounds studied, the absorption edge has a red-shift as the anion X changes from Cl to Br and to I and the cation changes from Pb to Sn when the organic framework A remains the same. As a result, CH3NH3SnI3 has the smallest optical gap and can respond to a very wide spectra of light. Our calculations of electronic properties of these compounds show that the fundamental band gap, consistent with the optical gap, is closely related to the cation and anion components. The variation of band gap with the component can be understood by the relative electronegativity between cation B and anion X when the organic framework A is unchanged. Our study not only highlights the important role of CH3NH3SnI3 for dye-sensitized solar cells, but also provides deep insight into the material engineering of ABX3 compounds.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 |