{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lattice stabilities, mechanical and thermodynamic properties of Al3 Tm and Al3 Lu intermetallics under high pressure from first-principles calculations
[Zhang Xu-Dong1, †,  , Jiang Wei1, 2]
, Jiang Wei1, 2]
, Jiang Wei1, 2]
|
|


† Corresponding author. E-mail:
Project supported by the Scientific Technology Plan of the Educational Department of Liaoning Province and Liaoning Innovative Research Team in University, China (Grant No. LT2014004) and the Program for the Young Teacher Cultivation Fund of Shenyang University of Technology, China (Grant No. 005612).
The effects of high pressure on lattice stability, mechanical and thermodynamic properties of L12 structure Al3Tm and Al3Lu are studied by first-principles calculations within the VASP code. The phonon dispersion curves and density of phonon states are calculated by using the PHONONPY code. Our results agree well with the available experimental and theoretical values. The vibrational properties indicate that Al3Tm and Al3Lu keep their dynamical stabilities in L12 structure up to 100 GPa. The elastic properties and Debye temperatures for Al3Tm and Al3Lu increase with the increase of pressure. The mechanical anisotropic properties are discussed by using anisotropic indices AG, AU, AZ, and the three-dimensional (3D) curved surface of Young’s modulus. The calculated results show that Al3Tm and Al3Lu are both isotropic at 0 GPa and anisotropic under high pressure. In the present work, the sound velocities in different directions for Al3Tm and Al3Lu are also predicted under high pressure. We also calculate the thermodynamic properties and provide the relationships between thermal parameters and temperature/pressure. These results can provide theoretical support for further experimental work and industrial applications.
The Al3M trialuminide compounds with L12 structure in aluminum alloys have attracted a great deal of attention recently due to their particularly attractive characteristics including low densities, high specific strengths, very high melting points and excellent oxidation resistances.[1–6] Among them, Al3Sc is the most studied intermetallics so far, since it has a thermodynamically stable L12 structure, relatively large volume fraction, and modest coarsening rate.[1,7,8] However, the high cost of Sc limits its industrial applications. Like Sc, Tm and Lu are cheaper than Sc and can also form thermodynamically stable L12-structured trialuminide compound.[9,10] The microstructure and mechanical properties of Tm and Lu containing Al alloys have been investigated, and it has been found that Tm and Lu can improve the creep-resistance properties of Al alloys by forming L12 structure Al3Tm and Al3Lu precipitates or solid solved in the matrix.[11] The recent studies of L12 structures Al3Tm and Al3Lu mainly focused on the electronic structure and elastic constant under ground states. Tao et al.[12] calculated the density of states and elastic modulus at the pressure of 0 GPa using first-principles calculations. Tsvyashchenko et al.[9] employed the time-differential perturbed angular correlations technique (TDPAC) to measure the parameters of hyperfine interactions in known Al3RE compounds. However, to the best of our knowledge, no first-principles calculations about the relationship between pressure and lattice stability, mechanical and thermodynamic properties for L12 structure Al3Tm and Al3Lu have been performed so far. So the effects of high pressure on phase stability, elastic constants and thermodynamic properties should be discussed for the further considerations. With the development of computer science and technology, first-principles calculations have successfully predicted the physical and chemical properties of the materials.[13–21] In the present work, the first-principles calculations are performed to better clarify and understand the lattice stabilities, mechanical and thermodynamic properties for L12 structures Al3Tm and Al3Lu under high pressure.
The first-principles calculations are based on density functional theory (DFT) and employ the plane-wave pseudo-potential total energy method as implemented in the VASP code.[22] Projector augmented wave (PAW) functions were used and exchange correlation functional was treated using the generalized gradient approximations (GGA) of the Perdew–Burke–Ernzerhof (PBE). The k-point meshes for Brillouin zone sampling were constructed using the Monkhorst–Pack scheme. The 12×12×12 k-point meshes were used for Al3Tm and Al3Lu. Spin polarization was considered in all calculations. The kinetic cut-off energy of 500 eV has been tested to be sufficient for convergence. The convergence of the energy was less than 1.0×10−5 eV. Force was converged to less than 1.0×10−4 eV·Å−1. In order to obtain the vibrational properties, we carried out the phonon calculation by using the supercells approach and used the 3×3×3 supercells for Al3Tm and Al3Lu. Real-space force constants were calculated using the density functional perturbation theory (DFPT) with the VASP code. Phonon frequencies were calculated by using the PHONOPY code.[23]
In the present work, L12 structure Al3Tm and Al3Lu belong to face-centered cubic crystal structures, each with space group Pm
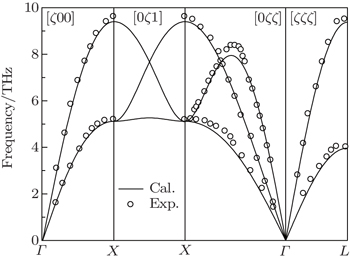
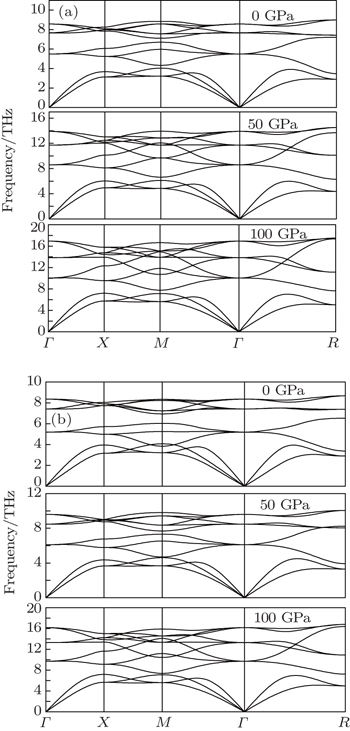
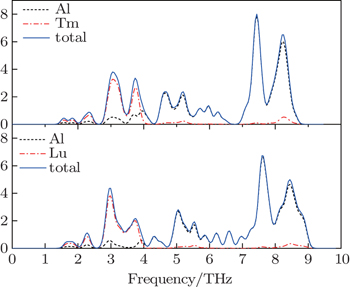
The vibrational properties play an important role in determining the physical properties of solids, such as dynamical stabilities, phase transitions, electron–phonon interactions and heat conductions and so on. We calculate the phonon spectra of Al3Tm and Al3Lu and examine their dynamical stabilities. The phonon spectrum curves at arbitrary values of wave vector q of the Brillouin zone (BZ) for Al, Al3Tm and Al3Lu are shown in Figs.
 | Fig. 1. Phonon dispersion curves of Al and the circles correspond to the experimental data. |
 | Fig. 2. Phonon dispersion curves for Al3Tm (a) and Al3Lu (b) under different pressures. |
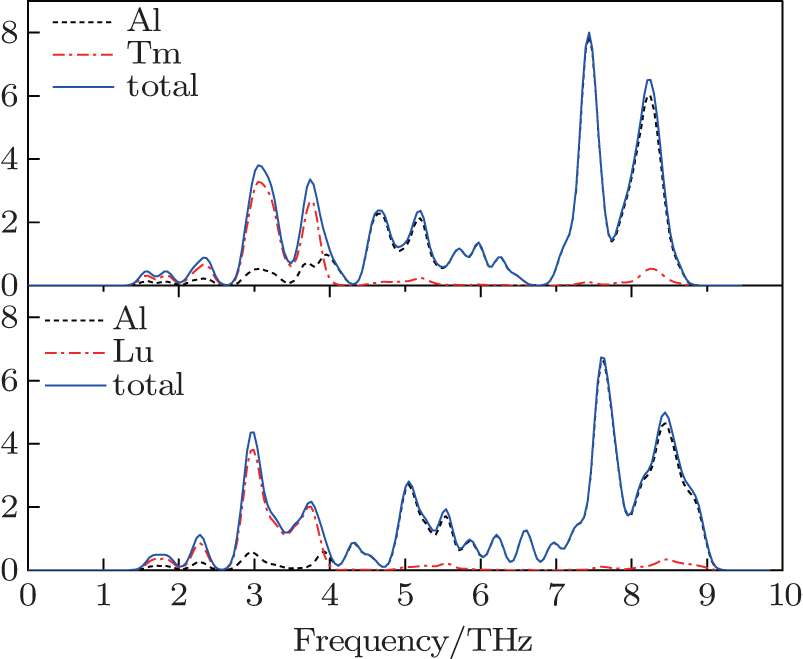 | Fig. 3. Calculated total and partial densities of phonon states for L12 Al3Tm, and Al3Lu at 0 GPa. |
| Table 1. Phonon frequencies (in THz) at the Γ point for L12 Al3Tm, and Al3Lu at different pressures (in GPa). . |
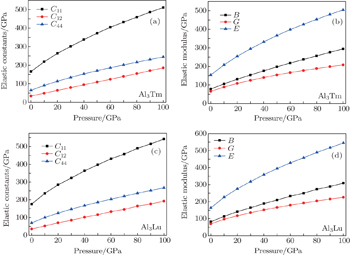
For elastic constants, we employ the stress-strain method proposed by Shang et al.[27] to calculate the elastic constants and use the Voigt–Reuss–Hill method[28] to obtain the Bulk modulus B, Young’s modulus E and shear modulus G. The value of bulk modulus (in GPa) B = (BV+BR)/2, where BV and BR are the Voigt’s and the Reuss’s bulk modulus respectively. The value of shear modulus G = (GV + GR)/2, where GV and GR are the Voigt’s and the Reuss’s shear modulus respectively. They are defined respectively as
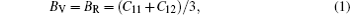
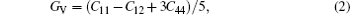
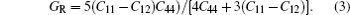
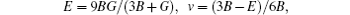
| Table 2. Elastic constants and elastic moduli (in GPa) of L12 Al3Tm, and Al3Lu at different pressures (GPa). . |
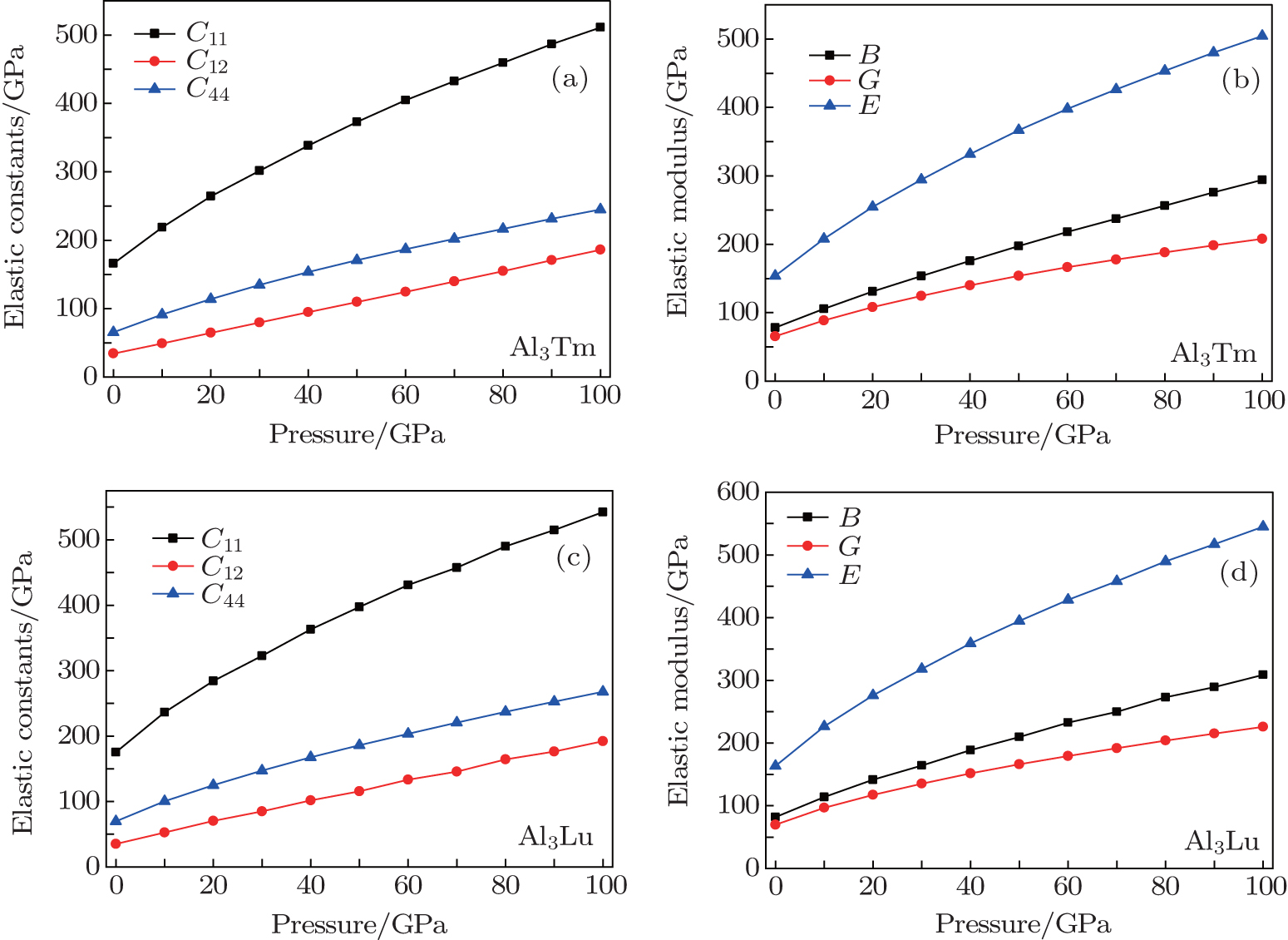 | Fig. 4. Elastic constants and elastic moduli of Al3Tm and Al3Lu at different pressures. |
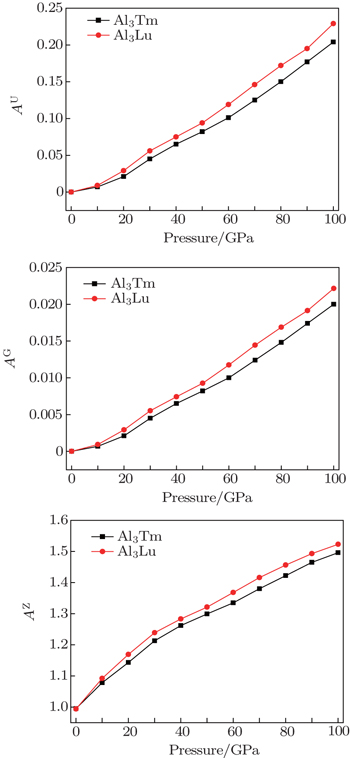
The elastic anisotropy constant is an important parameter of material and has many industrial and technological applications. Based on calculated elastic constants and moduli, we employ three methods to estimate the anisotropies for L12 Al3Tm, and Al3Lu. Zener factor AZ, Chung–Buessem anisotropy index AG, and universal elastic anisotropy index AU have been proposed as follows:[30–32]
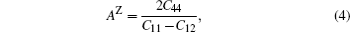
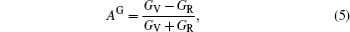
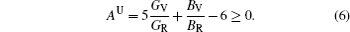
The calculated dependences of anisotropic factors (AZ, AG, AU) on pressure for Al3Tm and Al3Lu are shown in Fig.
 | Fig. 5. Pressure dependences of anisotropic factors (AU, AG, AZ) for Al3Tm and Al3Lu. |
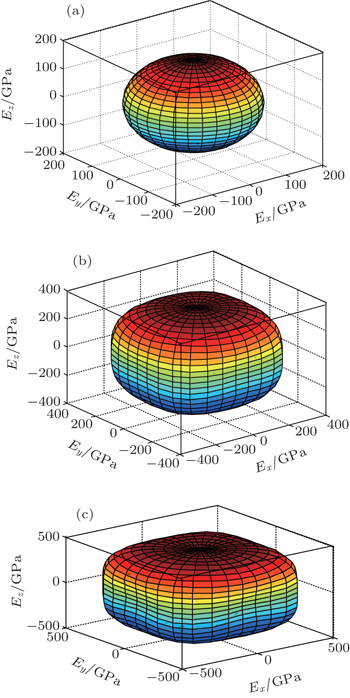
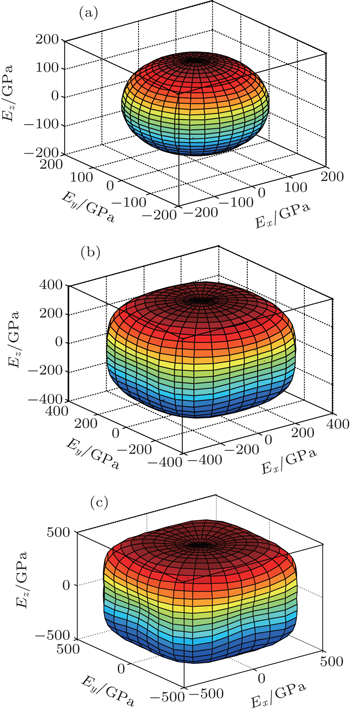
In order to further describe the elastic anisotropies for Al3Tm and Al3Lu, we plot the three-dimensional (3D) curved surface of Young’s modulus E in a given direction. The surface of 3D Young’s modulus E can be determined by using the following equation:[33]
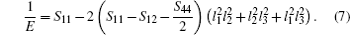
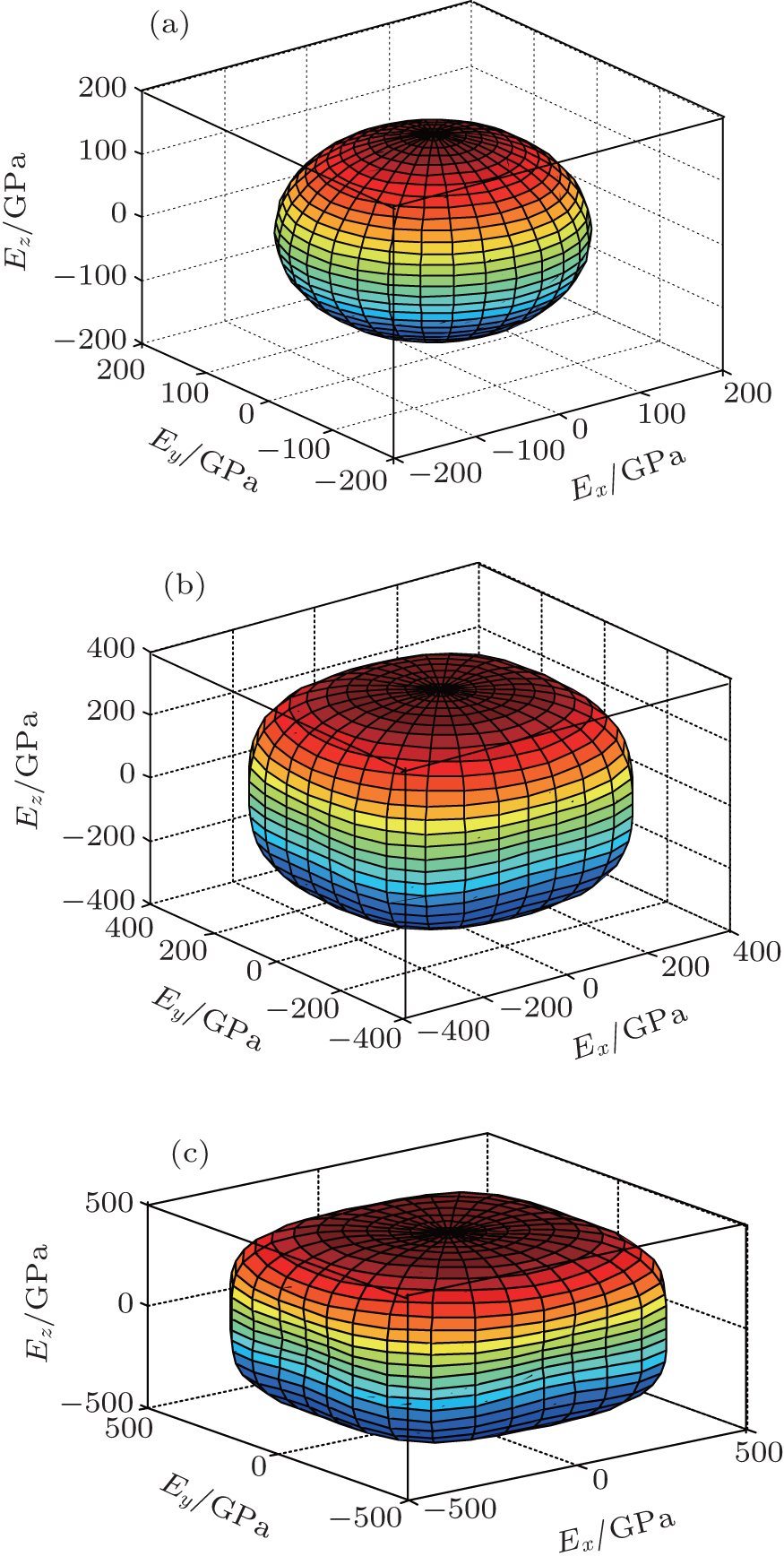 | Fig. 6. Directional dependences of Young’s modulus for Al3Tm at 0 GPa (a), 50 GPa (b), and 100 GPa (c), the axes are in units of GPa. |
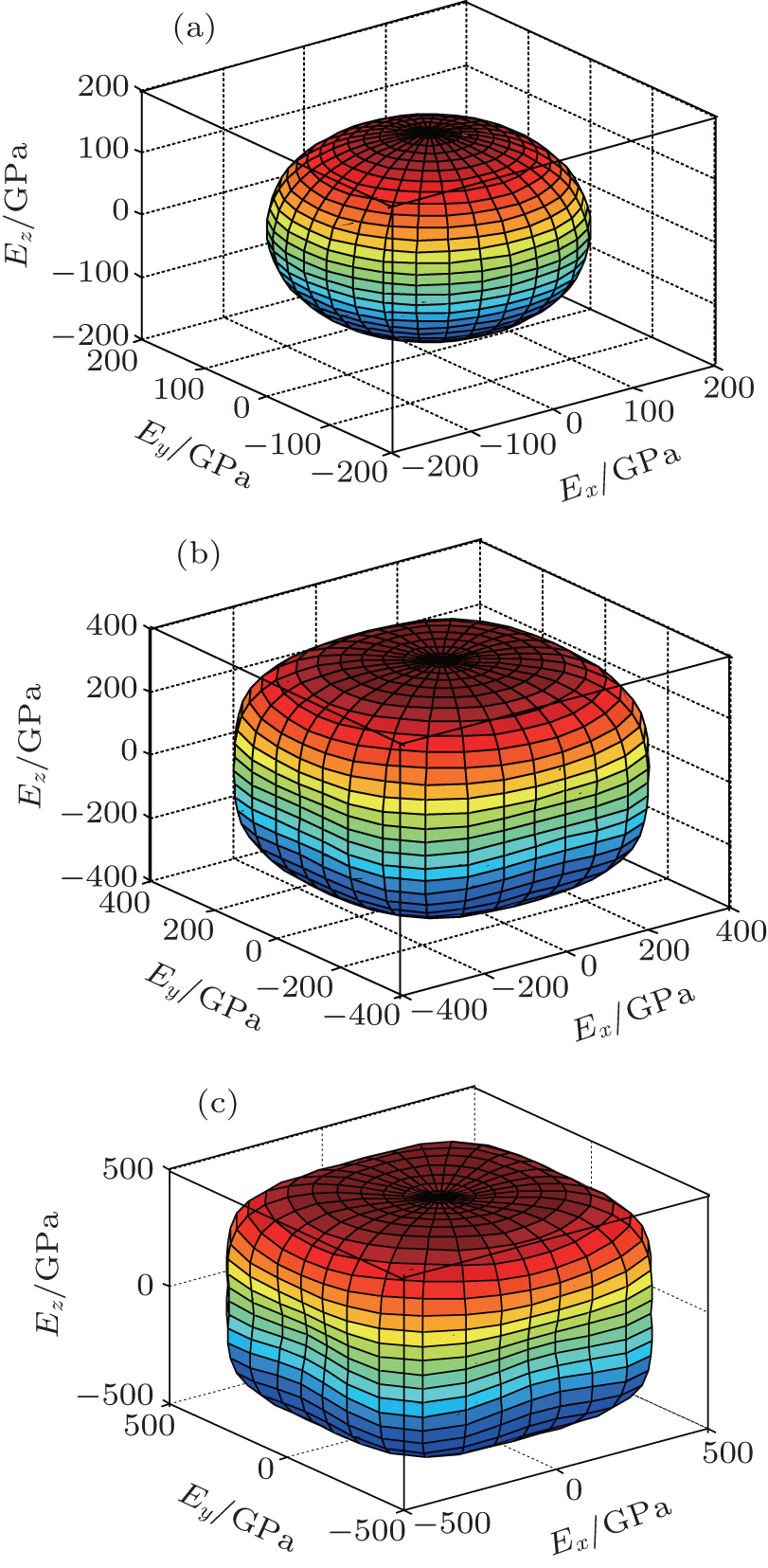 | Fig. 7. Directional dependences of Young’s modulus for Al3Lu at 0 GPa (a), 50 GPa (b), and 100 GPa (c), the axes are in units of GPa. |
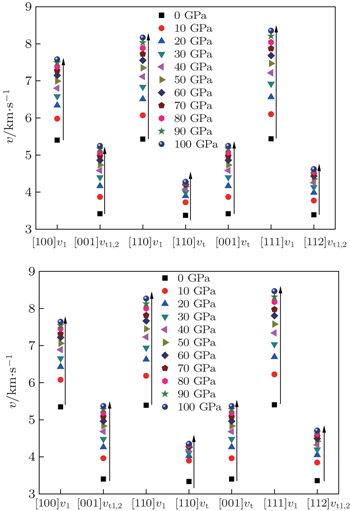
The sound velocities in solids are related to their elastic constants, thermal conductivities, and phonon frequencies. For L12, Al3Tm, and Al3Lu, we calculate the sound velocities of longitudinal (vl) and transverse waves (vt) along the [100], [110], and [111] crystal directions. We calculate sound velocities by using the following equations:[34,35]
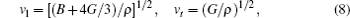
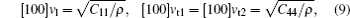
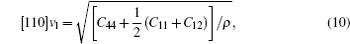
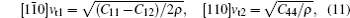
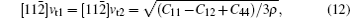
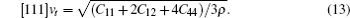
| Table 3. Sound velocities along different directions (km/s) and for different values of ρ (g/cm3) for L12, Al3Tm, and Al3Lu at different pressures (GPa). . |
The calculated sound velocities along three directions of Al, Al3Tm, and Al3Lu are presented in Table
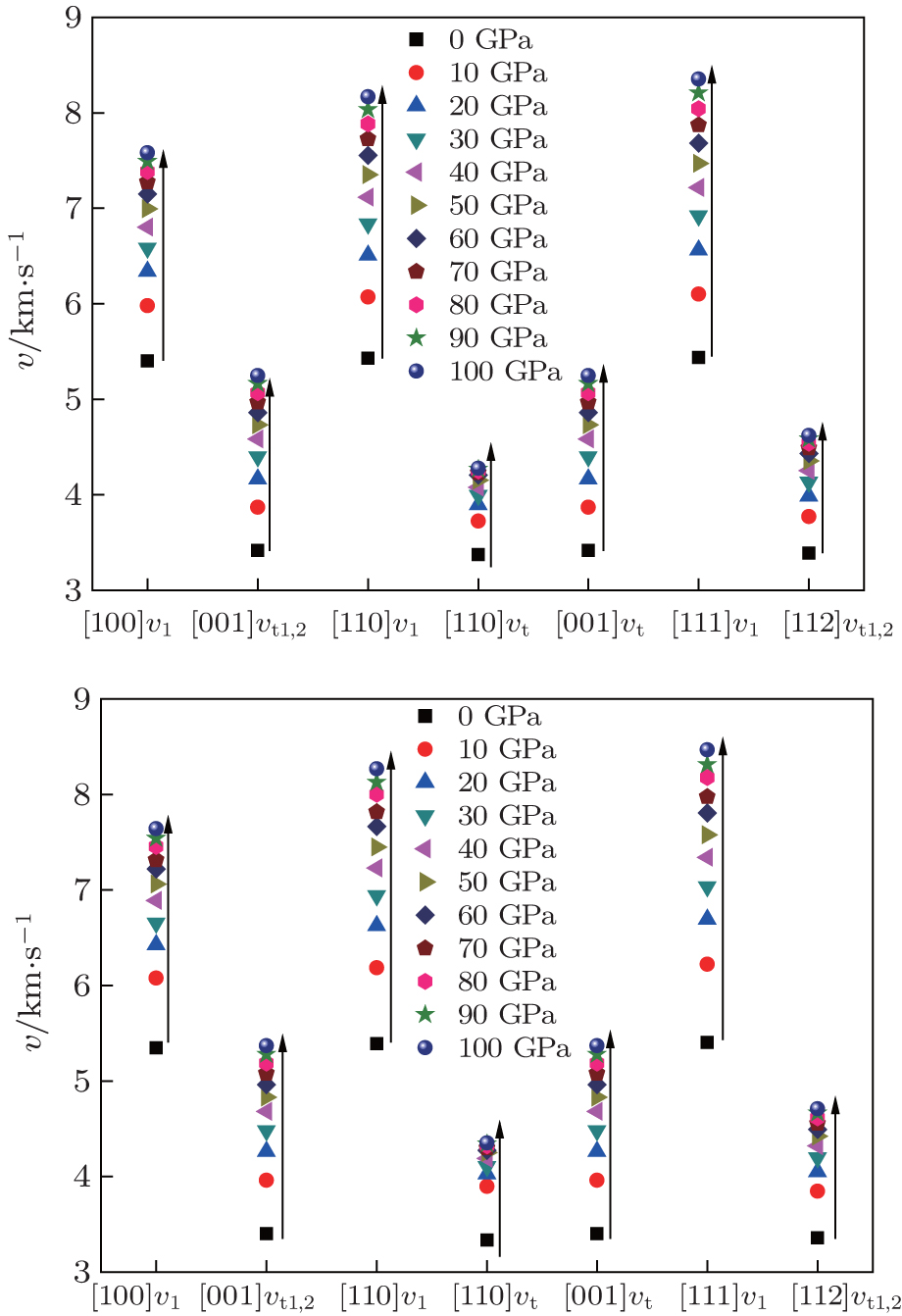 | Fig. 8. Sound velocities along different directions for Al3Tm (a) and Al3Lu (b), each as a function of pressure. |
The Debye temperature is an important parameter describing the material thermodynamic properties in solid state physics. We calculate the Debye temperatures of Al3Tm and Al3Lu by using the average sound velocities from the following equations:[37]
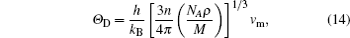
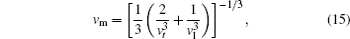
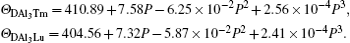
We can note that the values of zero-pressure Debye temperature ΘD are 411 K and 404 K for Al3Tm and Al3Lu respectively. The errors between the fitting ΘD and calculated ΘD in Table
| Table 4. Elastic wave velocities (m/s) and the Debye temperatures (K) for Al, Al3Tm, and Al3Lu. . |
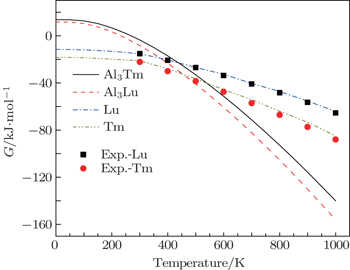
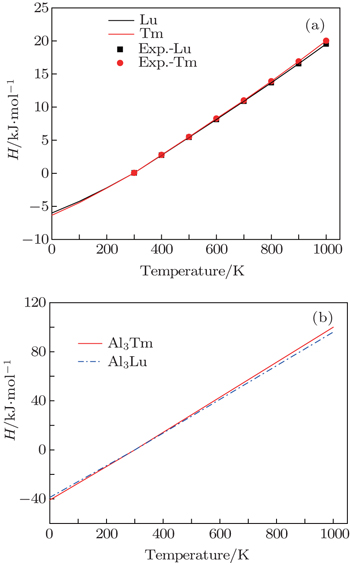
We calculate the thermodynamic properties of Al3Tm and Al3Lu by using the quasi-harmonic approximation (QHA) theory. In order to verify the correctness of the computational method, we calculate the values of Gibbs free energy (G) and enthalpy (H) of pure Tm, Lu, Al3Tm, and Al3Lu at 0 GPa. In Figs.
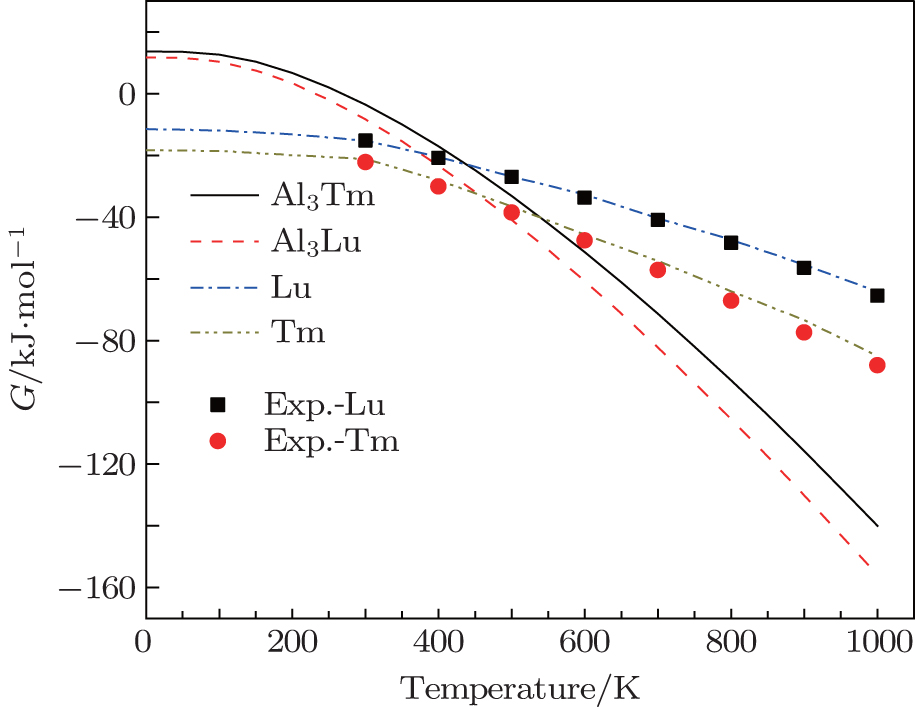 | Fig. 9. Temperature dependences of Gibbs free energy for Tm, Lu, Al3Tm, and Al3Lu at 0 GPa. |
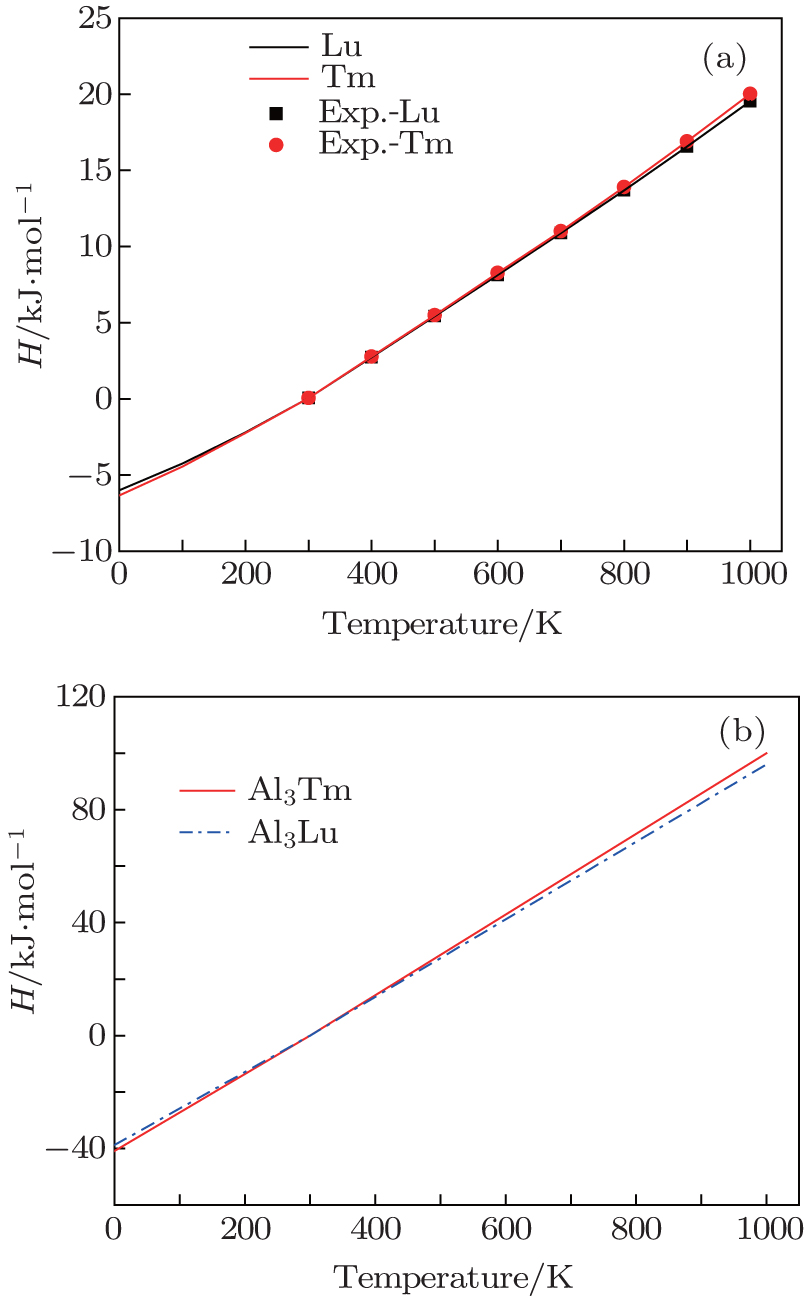 | Fig. 10. Temperature dependences of enthalpy for Tm, Lu (a), Al3Tm and Al3Lu (b). |
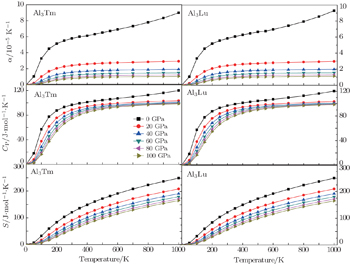
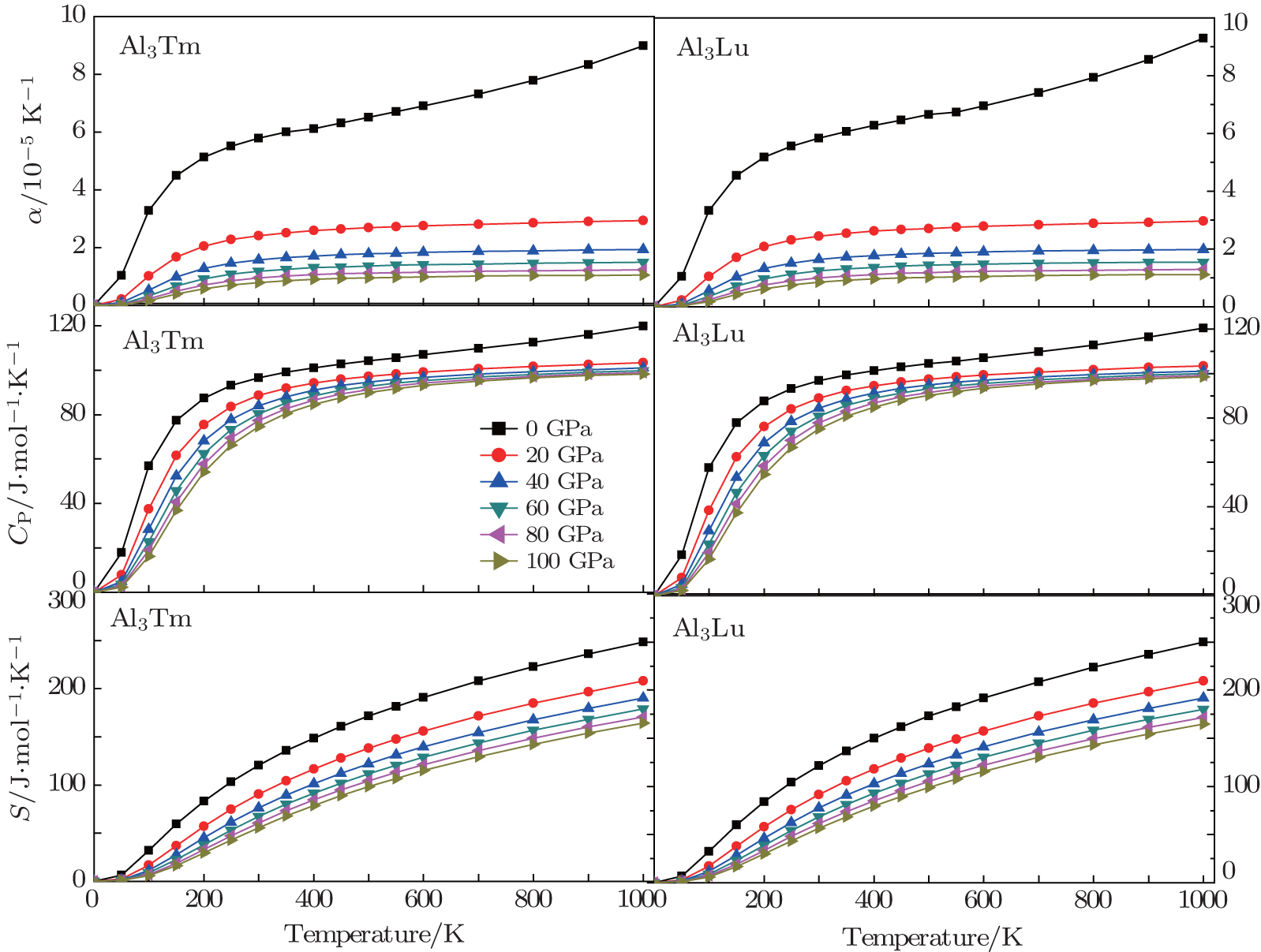 | Fig. 11. Temperature dependences of thermal parameters for Al3Tm and Al3Lu. |
It indicates that the increasing of pressure influences the increase of CP. The curves of entropy S at different pressures are shown in Fig.
In this work, the dynamic stabilities, elastic and thermodynamic properties of Al3Tm and Al3Lu under high pressure are investigated by using first-principles calculations. According to the vibrational properties, we can predict that Al3Tm and Al3Lu will keep the lattice stable up to 100 GPa. The elastic constants and elastic moduli for Al3Tm and Al3Lu indicate that the two compounds keep mechanically stable and possess a brittle nature in the considered pressure range. The calculated anisotropic constants AG, AU, and AZ and 3D curved surface of Young’s modulus show that Al3Tm and Al3Lu are both isotropic at 0 GPa and are anisotropic under high pressure. The sound velocities in different directions and Debye temperatures under high pressure are also calculated for both phases. We also calculate the thermodynamics properties by the QHA method and discuss the relationships between these thermal parameters and pressure as well.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 |