{kind=link}
{kind=link}
{kind=link}
High-pressure structural properties of tetramethylsilane
[Qin Zhen-Xing1, 2, Chen Xiao-Jia3, †,  ]
]
]
 |


† Corresponding author. E-mail:
Project supported by the Cultivation Fund of the Key Scientific and Technical Innovation Project from Ministry of Education of China (Grant No. 708070), the Fundamental Research Funds for the Central Universities, South China University of Technology (Grant No. 2014ZZ0069), the National Natural Science Foundation of China (Grant No. 51502189), and the Doctoral Project of Taiyuan University of Science and Technology, China (Grant No. 20132010).
High-pressure structural properties of tetramethylsilane are investigated by synchrotron powder x-ray diffraction at pressures up to 31.1 GPa and room temperature. A phase with the space group of Pnma is found to appear at 4.2 GPa. Upon compression, the compound transforms to two following phases: the phase with space groups of P21/c at 9.9 GPa and the phase with P2/m at 18.2 GPa successively via a transitional phase. The unique structural character of P21/c supports the phase stability of tetramethylsilane without possible decomposition upon heavy compression. The appearance of the P2/m phase suggests the possible realization of metallization for this material at higher pressure.
As an important challenge in modern physics and astrophysics, the metallization of hydrogen has long been a major driving force in high-pressure science and technology development,[1] which originates from the possible superconductivity with high Tc(> 200 K) under sufficiently strong compression.[2,3] Recently, several independent measurements indicate that metallic hydrogen has not been reached yet even at 360 GPa,[4,5] whereas much higher pressures (e.g., > 400 GPa) are thought to be needed for metallization,[6] which currently are a great challenge for hydrogen with high-pressure techniques. In 2004, Ashcroft already with great interest suggested that hydrogen-dominant hydrides could also be high-temperature superconductors in monatomic and molecular phases, providing an alternative way to achieve the metallic hydrogen. By this argument, such covalent hydrides could exhibit metallization at significantly reduced pressures compared with pure hydrogen due to chemical precompression.[7] The recent breakthrough in the discovery of superconductivity above record high 190 K in H–S compound[8] offers convincing evidence for this idea. Group IVa hydrides were specifically suggested as potential candidates for this material and many experimental and theoretical efforts are currently underway to investigate this prediction, such as SiH4,[9–18] GeH4,[19–23] SnH4,[24–27] and PbH4.[28] However, very recent experiments show the possible decomposition of SiH4 under irradiations from x-ray and lasers,[29,30] which results in a loss of superiority for these compounds to further investigate the metallization of bulk hydrogen. For achieving the metallic hydrogen at “low” pressure, it is extremely urgent to search other hydrogen-rich compounds on the group IVa hydrides.
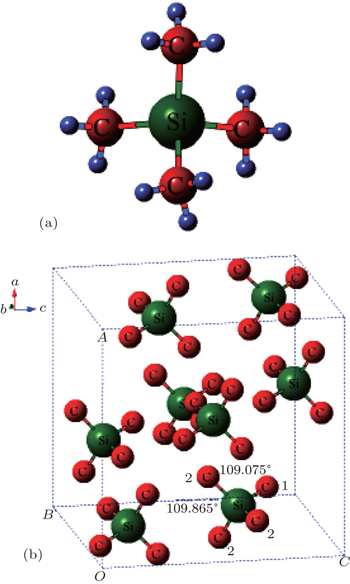
Tetramethylsilane (TMS), Si(CH3)4, has spurred tremendous interest in the spectrum due to its highly symmetrical characters.[31–36] In the molecular structure in Fig.
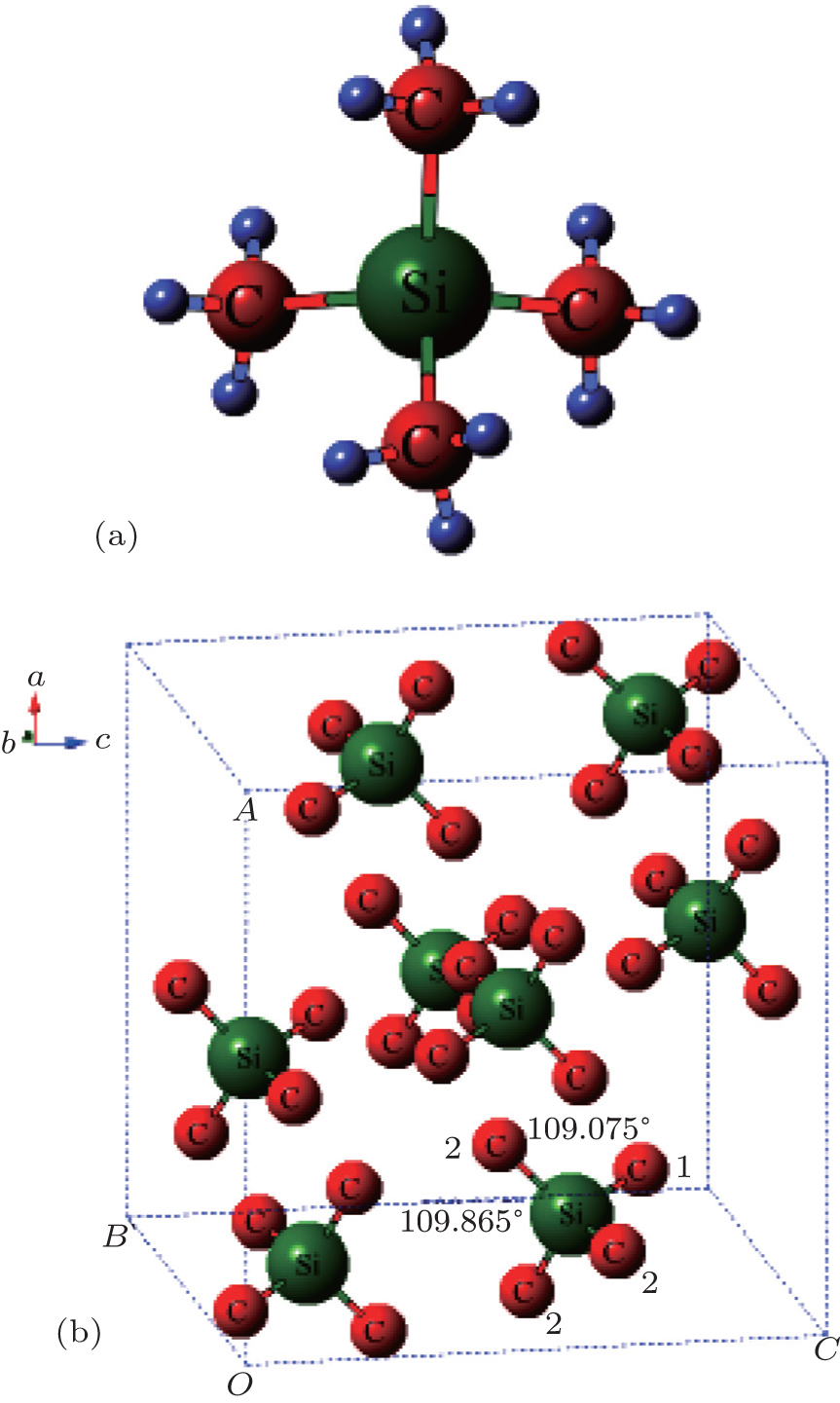 | Fig. 1. (a) Configurations of TMS with respect to ideally tetrahedral 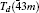  |
On the basis of the global minimization of the lattice energy, several competitive crystal structures have been proposed for the phases of TMS in recent computational studies,[37] while there are only a few experimental data for its molecular and crystal structures even at lower pressures.[38,40] Such information is important for understanding the evolution in the electronic structure leading to metallization. In this paper, we present a high-pressure study of TMS by synchrotron powder x-ray diffraction (XRD) at up to 31.1 GPa. We observe a similar process of phase transitions although it shows slight hysteresis compared with the results from Raman spectrum. TMS underwent Pnma → P21/c → P2/m structural transitions via 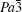
TMS (m.p. 178 K, b.p. 299 K) as transparency liquid with 99.9% purity was purchased from Alfa-Aesar and used without further purification. The high-pressure experiments for TMS were carried out by using Diamond anvil cell (DAC) with beveled anvils and the culets of 300 μs. A hole of ∼ 100 μs in diameter, drilled in a preindented tungsten gasket, served as the sample chamber. To avoid volatilizing, the bottom of DAC was put into ice-water mixture half an hour before loading the sample. Liquid TMS was loaded into the chamber of DAC with a syringe. Because of liquid sample, no pressure medium was used and ruby grains had been placed previously as pressure marker. Considering the volatilizing of samples, pressure was increased to 0.6 GPa after loading well the samples. Synchrotron radiation measurements were performed at the X17C beamline of National Synchrotron Light Source at Brookhaven National Laboratory via angle-dispersive diffraction techniques by using monochromatic radiation λ = 0.3989 Å. The sample-to-detector distance and the image plate orientation angles were calibrated by using CeO2 standard. The two-dimensional (2D) diffraction images were converted to the plots of 2θ versus intensity by the FIT2D software.
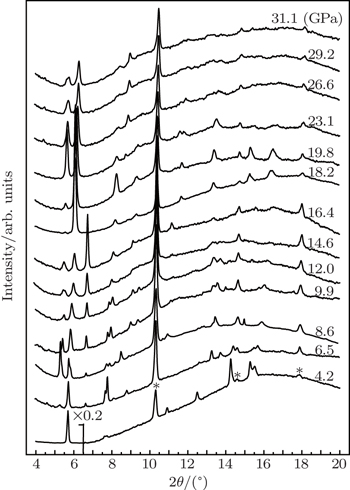
Synchrotron powder x-ray diffraction measurements on TMS are performed to determine detailed structural information for the possible phases under pressure. Figure
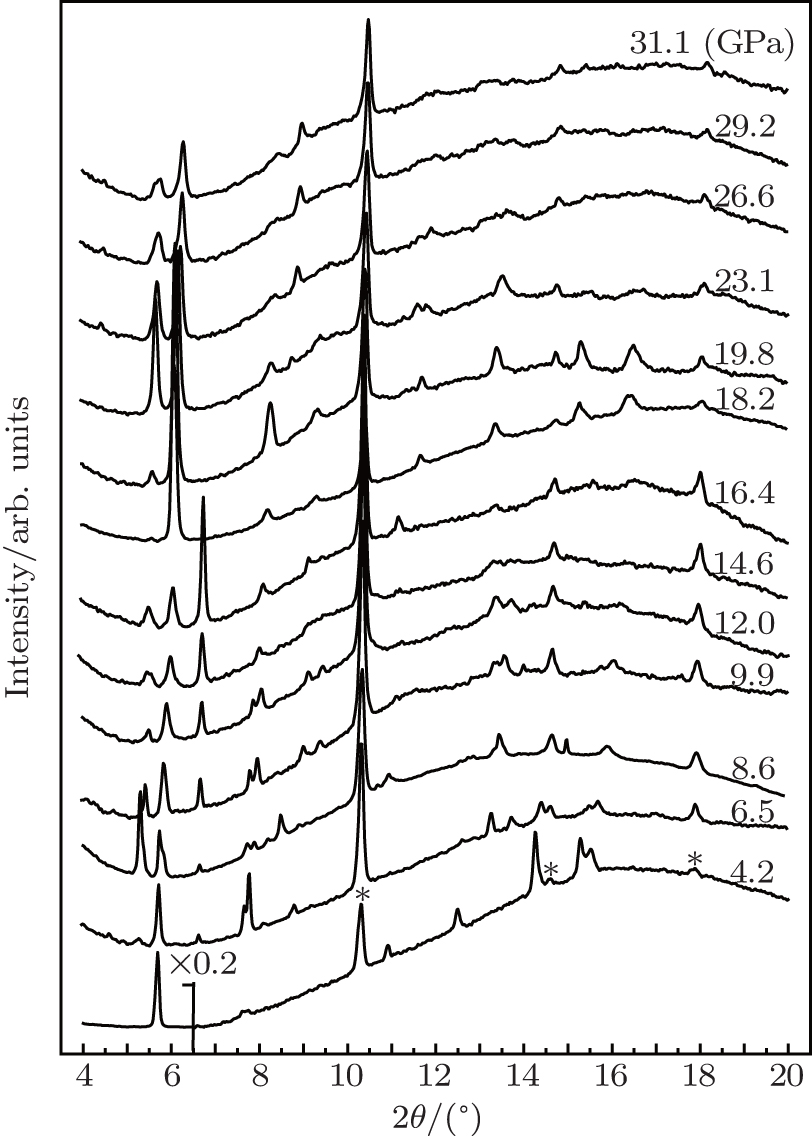 | Fig. 2. Synchrotron radiation x-ray (λ = 0.3989 Å) diffraction patterns of TMS during the pressurization from ambient pressure to 31.1 GPa. Red asterisks marked indicate the signals from the gasket material tungsten. The intensity of first peak is reduced to 1/5 at 4.2 GPa due to strong preferred orientation of (211) in Pnma phase. |
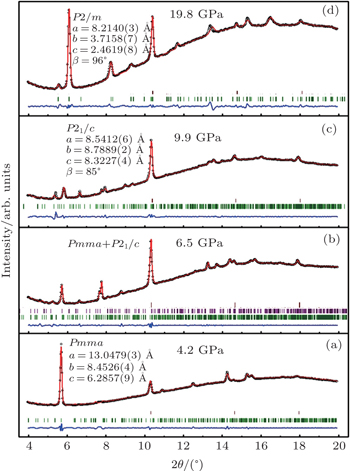
To investigate the crystal structure of each phase, the diffraction patterns obtained at selected pressures are indexed using Dicvo104 and refined using the Le Bail method with GSAS software.[41] For TMS, it is reported that TMS crystallizes into cubic structure with the space group 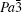
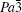
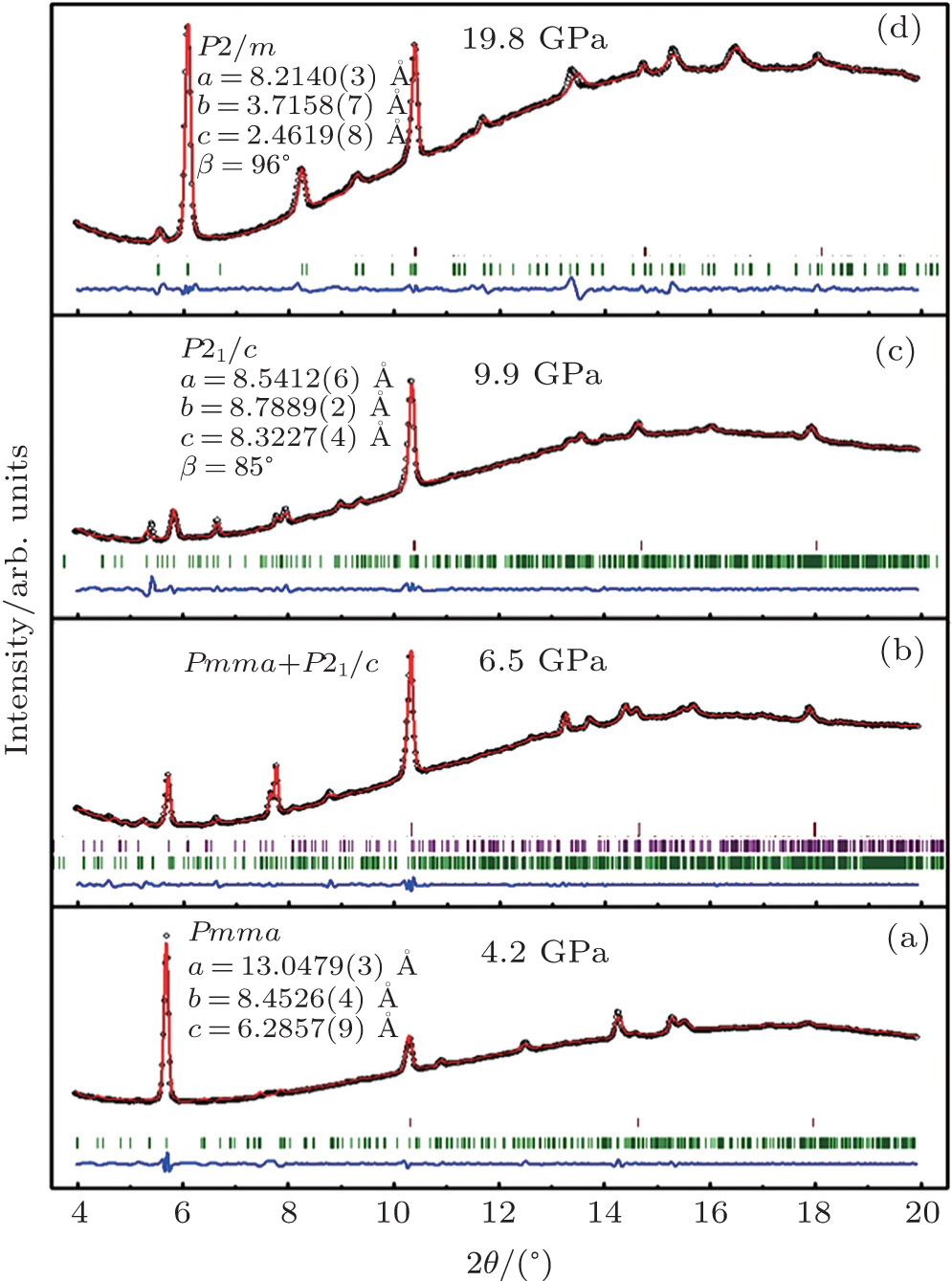 | Fig. 3. X-ray powder diffraction patterns of liquid TMS at pressures of (a) 4.2, (b) 6.5, (c) 9.9, and (d) 19.8 GPa, respectively. The refined lattice parameters for the corresponding space groups are given, respectively. The open circles represent the measured intensities and the red lines represent the results of profile refinements by the best LeBail-fit with each space group. The positions of the Bragg reflections are marked by vertical lines Pnma is shown as green and P21/c is shown as purple in panel (b) and the difference profiles are shown at the bottoms (blue lines). |
For phase IV of TMS at 9.9 GPa, all peaks from the XRD pattern are indexed mainly to the monoclinic system. However, it is difficult to determine the space group for this new phase, whereas P21/c is a candidate because tetrahalides of the group IVa elements, MX4 (M = Si, Ge, Sn; X = Cl, Br) with halogen atoms each have a comparable size to a methyl group crystallized in P21/c.[45–50] Considering the Raman results that CH3 groups are locked in position and the whole groups move like one atom,[39] the P21/c space group will be the most reasonable solution to the structure of TMS at phase IV. In addition, the P21/c space group is also predicted as the third best structure of TMS energetically and appears repeatedly with increasing the energy of crystal structure.[37] Figure
There is no information about crystal structure available at the highest-pressure phase (phase V) of TMS in current work. The diffraction patterns yield several orthorhombic and monoclinic systems at 19.8 GPa with reasonable lattice parameters. According to the study of high-pressure Raman spectrum,[39] phase V should possess lower symmetry because the number of Raman bands greatly increases in the phase,[51] thus the orthorhombic systems have been ruled out. Furthermore, it is quite interesting that all monoclinic cells are characterized by different lattice parameters and the same space group P2/m at this pressure. It might be the most reasonable of the lattice parameters a = 8.2042 Å, b = 3.7287 Å, c = 2.4630 Å, and β = 97.714° with a volume of 74.66 Å3 because of remarkable value of the figures of merit (M, F) at this pressure. Figure
The R values are Rp = 0.2%, Rwp = 0.4% for the fitting at 4.2 GPa, Rp=0.1%, Rwp=0.3% at 6.5 GPa, Rp = 0.3%, Rwp = 0.4% for the fitting at 9.9 GPa, and Rp = 0.4%, Rwp = 0.5% at 19.8 GPa.
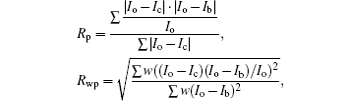
Although the use of the Le Bail method can give reasonable space groups and yields accurate lattice parameters of the structures, this method has not provided any information about the internal coordinates. Since the texture of the sample combined with the extraordinarily large hydrogen content presents limitation to meaningful Rietveld structural refinements, as a substitution, we compare the crystal characteristic of TMS at phase IV with that of the typical hydrogen-rich compound SiH4. The reason is that the structure P21/c is the last one for SiH4 existing before becoming partial decomposition and amorphization or polymerization upon compression despite it is still an insulating molecular solid,[30] which is helpful for us to understand the procedure of structural change on TMS.
For SiH4 as shown in Table
| Table 1. Structural characteristics of TMS phase IV compared with those of tetrahalidies having the SnBr4-type structure. . |
Recently, our work on the structure of Sn(CH3)4 (TMT) by synchrotron x-ray diffraction measurement has shown to crystallize at 12.5 GPa into space group P2/m, which yields lattice parameters of a = 11.8438 Å, b = 6.3417 Å, c = 2.9825 Å, and β = 94° (Z = 2).[61] TMS also confirms the space group P2/m with the lattice parameters of a = 8.2140 Å, b = 3.7158Å, c = 2.4619 Å, and β = 96° (Z = 2) at phase V via P21/c space group upon compression. For the optimal calcuation on P2/m structure of TMT, the results show the metallic character with large dispersion bands crossing the Fermi level although our optical observations seemingly do not support the metallization in TMT,[61] which could be a indication to undergo a metallic state for TMS and TMT soon with increasing pressure. Excitingly, TMS does yield a possible semimetallic state with the appearing of new and softening vibrational modes at 96 GPa,[39] which resembles the case on hydrogen in phase IV upon compression to 278 GPa, indicated by Raman spectrum.[4] These theoretical and experimental results are encouraging for the experimental detection of the metallization in such materials because the resistance measurements on a similar aromatic hydrocarbon indeed reveal the metallic state at a much low pressure of 40 GPa than generally thought.[62] Further electrical transport measurements are expected on the studied material.
We perform high-pressure powder x-ray diffraction measurements on TMS at up to 31.1 GPa. Our results reveal the homogeneous process of phase transitions for TMS with compression to 31.1 GPa compared with the aforementioned work on Raman spectrum investigations of liquid TMS. A new phase possesses a space group of Pnma at 4.2 GPa, which is identified by the XRD patterns at pressure up to 31.1 GPa. After that, the material transforms into phase IV with the P21/c symmetry and phase V with monoclinic P2/m structure at the corresponding pressures of 9.9 GPa and 19.8 GPa respectively via a transitional phase in a pressure range of 6.5 GPa–8.6 GPa. These structural transitions suggest that they result from the changes in the inter- and intra-molecular bonding of this material. Interestingly, P21/c structure of TMS has unique lattice parameters compared with those of the compounds containing relevant tetraheddral molecules, which makes TMS continue to crystallize but not realize decomposition nor amorphization upon compression. Additionally, the appearing of P2/m space gronp in phase V offers a possibility to achieve the metallization in this material with further compression. Such structural information will encourage one to experimentally detect the metallization in such materials by other methods.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 | |
| 52 | |
| 53 | |
| 54 | |
| 55 | |
| 56 | |
| 57 | |
| 58 | |
| 59 | |
| 60 | |
| 61 | |
| 62 | |
| 63 |