{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Modeling the temperature-dependent peptide vibrational spectra based on implicit-solvent model and enhance sampling technique
[Wu Tianmin 1 , Wang Tianjun 2 , Chen Xian 2 , Fang Bin 2 , Zhang Ruiting 2 , Zhuang Wei 2, †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant No. 21203178), the National Natural Science Foundation of China (Grant No. 21373201), the National Natural Science Foundation of China (Grant No. 21433014), the Science and Technological Ministry of China (Grant No. 2011YQ09000505), and “Strategic Priority Research Program” of the Chinese Academy of Sciences (Grant Nos. XDB10040304 and XDB100202002).
We herein review our studies on simulating the thermal unfolding Fourier transform infrared and two-dimensional infrared spectra of peptides. The peptide–water configuration ensembles, required forspectrum modeling, aregenerated at a series of temperatures using the GB OBC implicit solvent model and the integrated tempering sampling technique. The fluctuating vibrational Hamiltonians of the amide I vibrational band are constructed using the Frenkel exciton model. The signals are calculated using nonlinear exciton propagation. The simulated spectral features such as the intensity and ellipticity are consistent with the experimental observations. Comparing the signals for two beta-hairpin polypeptides with similar structures suggests that this technique is sensitive to peptide folding landscapes.
Vibrational spectroscopy is a commonly used tool for studying protein conformation dynamics. [ 1 – 11 ] The amide I vibrational band, which mainly originates from the backbone C=O stretch vibrations, has different features for different secondary structure motifs of proteins, and is extensively used for protein configuration detection. [ 12 – 18 ] The two-dimensional infrared (2DIR) spectra of peptide further improve the resolution of the local conformational changes in the peptides; in 2DIR, the fundamental transitions are revealed by the diagonal peaks and the off-diagonal peaks probe the fluctuations of the correlations in the system. [ 18 – 27 ]
Thermal unfolding experiment, in which the peptide spectra are obtained at a series of temperatures, is employed to study the thermal stability of a given peptide structure. [ 27 – 32 ] Gursky et al . [ 33 ] monitored protein aggregation during thermal folding/unfolding by circular dichroism (CD) spectroscopy. Gai et al . [ 34 ] carried out the thermal unfolding IR and CD spectroscopic studies on polypeptides, and found that the turn structure plays a key role in peptide folding. Tokmakoff distinguished a folded state and a misfolded state for Trpzip2 peptide by combining 2DIR spectroscopy with thermal unfolding. [ 35 ]
Theoretical spectroscopy modeling, based on explicit solvent molecular dynamics (MD) simulations, has been extensively used to simulate and interpret complex and congested protein vibrational spectra. [ 23 – 28 ] Further, our recent studies suggested that comprehensive sampling of the peptide or protein configuration distribution helps improve the modeling of protein vibrational spectra. [ 36 – 39 ] It is, however, too expensive to carry out configuration sampling for proteins based on a converged explicit solvent model, even at a specific temperature. [ 40 – 43 ] It leads to multiplied computational cost for the thermal unfolding spectra because the sampling is needed for a series of temperatures. [ 38 , 39 ]
We modeled the thermal unfolding IR and 2DIR spectra of the peptides based on configuration distributions generated using implicit solvent MD simulation combined with the integrated tempering sampling (ITS) method. [ 38 , 39 , 44 , 45 ] Our method reproduces various experimental observables such as melting temperatures, [ 45 – 47 ] lineshapes of both linear and 2DIR spectra [ 48 ] and temperature dependences of the peak intensities and ellipticities of the isotope-labeled peaks. [ 48 ] Implicit-solvent-based simulation thus provides a cost-effective means to model temperature-dependent vibrational spectra of peptides. [ 45 ] In this paper, we first present the techniques we use, and then discuss the quality of simulation based on the comparison with the experimental IR and 2DIR signals of Trpzip2 and Trpzip4 peptides.
In the simulation, implicit-solvent MD simulation combined with ITS is used to generate the peptide configuration ensemble at a specific temperature T . A subgroup of thus-generated peptide configurations is then selected and soaked into the simulation box with explicit water molecules. Short simulations are then carried out to generate the trajectories needed for modeling the spectra. Herein, we briefly describe the techniques used in our simulation.
Unlike explicit-solvent models that are particularly time-consuming for large molecules, the implicit-solvent models represent the real solvent as a continuous medium whose properties can be described by different analytical functions. These treatments significantly improve computational efficiency and reduce errors in statistical averaging due to incomplete sampling of solvent conformations. [ 49 ] The generalized born (GB) implicit-solvent model approaches an approximate numerical solution of the Poisson–Boltzmann equation to describe the electrostatic energy. [ 50 ] The effective Born radius of each solute atom, which is the important parameter to calculate the solvation free energy, is given by the Coulomb field approximation [ 51 ]
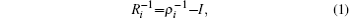
The ITS method, [ 44 ] developed by Gao et al ., is effective in improving the efficiency of sampling over a large energy range in MD simulations. The approach uses Boltzmann distribution functions at multiple temperatures to allow sampling of the configurations of the system over a wide energy range. [ 44 ]
Details of this technique have been extensively discussed elsewhere. [ 45–47 , 52 , 53 ] Here we briefly introduce the main concept: a generalized distribution function p ( U ) can be applied as a function of the potential energy of the system U as integration over β = ( k B T ) −1 , where k B is the Boltzmann constant and T is the temperature of the system [ 44 ]
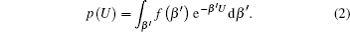
As running simulations on a modified potential U ′( U ) in an MD simulation, we can obtain the distribution function of Eq. (
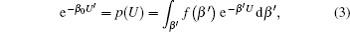
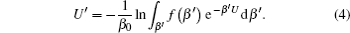
As demonstrated in Eq. (
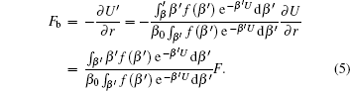
In Eq. (
Amide I unit (O=C–N–H) vibrations are localized on each residue along the backbone polypeptide bonds with no overlapping transition charge densities. [ 17 ] The Frenkel exciton model is therefore used to construct the amide-I fluctuating vibrational Hamiltonians for each snapshot along the trajectories: [ 54 , 55 ]
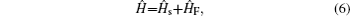
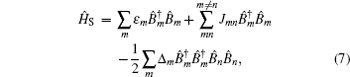
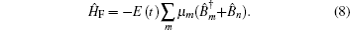
In these equations, Ĥ S is the system Hamiltonian and Ĥ F represents the interaction with the optical field, E ( t ). 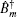
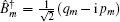

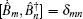
The third-order response function is calculated as follows: [ 23 , 58 – 60 ]

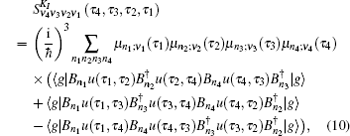
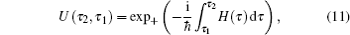
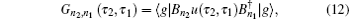
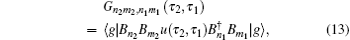
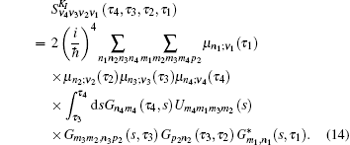
A time integral over the interval s between interactions with the
To save computational resources, Green’s function in Eq. (
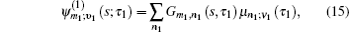
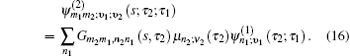
In Eq. ( 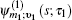
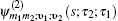
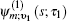
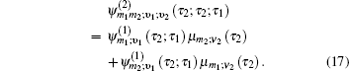
Equation (
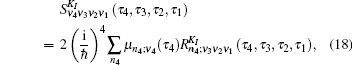
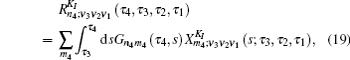
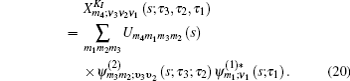
The one- and two-exciton wave functions are computed by direct integration of the Schrödinger equation. The one-exciton wave function is denoted as
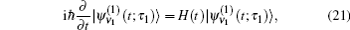
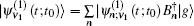
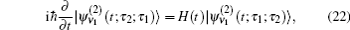
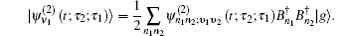
Since the features of the IR spectra of the amide I vibration, such as band position and line-shape, are strongly dependent on protein secondary structure, a variety of computational methods for numerically simulating amide-I vibrational spectra have been developed to simulate IR, VCD, and 2DIR spectra for a variety of polypeptideand protein secondary structures over the past decades. [ 17 , 20 , 55 , 61 – 68 ] Myshakina et al . [ 69 ] estimated the vibrational coupling strengths of amide-I modes of trialanines in right-handed α -helix and polyproline-II conformations using the isotopolog technology to isotope label 13 C= 18 O and ND or ND 2 in trialanines. Torii and Tasumi performed ab initio molecular orbital calculations for model glycine di- and tri-peptides with various ϕ and ψ angles to obtain the map of amide I vibrational coupling constants between the nearest peptide groups and those of the second-nearest peptide groups. [ 16 ] Furthermore, density functional theory (DFT) calculations for glycine dipeptide were performed by Hayashi and Mukamel to estimate the coupling constants between the neighboring polypeptide units for amide I, II, III, and vibrational modes. [ 70 ] The amide-I local modes for peptides are fully characterized using the eigenvectors of the four atoms (O=C–N–H) forming the peptide bond. Based on the full anharmonic vibrational Hamiltonian of the glycine di-peptide, they determined the inter-mode coupling constants, and used them to calculate the IR and VCD spectra of the α -helix model. These computational strategies considering the transition dipole–dipole interactions for calculating the amide-I mode coupling constants developed by Mukamel et al . are more accurate than the simple transition dipole coupling model. [ 69 , 70 ] Hamm, Woutersen, [ 71 ] and Jansen et al . [ 54 ] proposed an improved method to calculate the amide-I vibrational mode coupling between two local amide-I modes considering the transition charge–charge interactions. The transition-charge or dipole coupling models can successfully predict the long-range vibrational coupling between a pair of local amide-I modes; however, since transition charge coupling theory is strictly based on electrostatic interactions, it cannot fully demonstrate the through-bond coupling between neighboring polypeptides. Therefore, based on quantum chemistry calculations of model dipeptides by varying the dihedral angles ϕ and ψ , a series of amide-I vibrational-mode coupling constant maps were constructed, [ 16 , 54 , 56 , 72 , 73 ] and were used to describe the amide-I vibrational-mode couplings between pairs of nearest-neighboring local amide-I modes in polypeptides.
The frequencies of the amide-I local vibrational modes are required to construct the Frenkel vibrational exciton models. The local backbone conformation and electrostatic environment strongly influence the amide-I local mode frequencies. Mendelsohn et al . subtracted an ad hoc value associated with the strengths of hydrogen bonding interactions (calculated by ab initio methods) to estimate the amide-I local vibrational mode frequencies. [ 74 , 75 ] The amide I, II, III, and A local vibrational-mode frequencies were calculated from an electrostatic DFT map of NMA developed by Hayashi et al . [ 76 ] The Hessian matrix reconstruction method developed by Cho et al . [ 77 – 79 ] was used to calculate the fundamental-mode frequencies and the inter-mode coupling constants of polypeptides and nucleic acids, based on the results of ab initio vibrational analysis. The coupling constants and local-mode frequencies calculated by this method have been successfully applied to simulate the IR and 2D IR spectra.
To calculate the spectral signals, we randomly selected a tiny subset 
After the explicit-solvent solvation is done, the minimization, heating, and cooling were done, with all the atoms in the polypeptide fixed. [ 38 , 39 ] Finally, a short-time (10 ps) NPT ensemble equilibrium MD simulation, with no constraint on polypeptides was carried out for the whole system to generate a group of short trajectories, which were later used for further Hamiltonian construction and wave-function propagation. [ 38 , 39 ] We assume that during this short time the configuration of the polypeptide only changes slightly compared with the initial conformation. Considering the size and flexibility of the beta-hairpin peptides, this is a reasonable assumption. To simulate temperature-dependent infrared spectroscopy, this procedure was carried out for a series of temperatures from 285 to 350 K. [ 38 , 39 ]
The SPECTRON package [ 80 ] and the nonlinear exciton propagation (NEP) method [ 19 , 60 ] were used to calculate the spectra of polypeptides. The fluctuating vibrational Hamiltonians for the amide-I bond were constructed for each trajectory that were generated in previous NPT MD simulations, and then used to integrate the NEE equations. [ 23 ] The weight distribution 
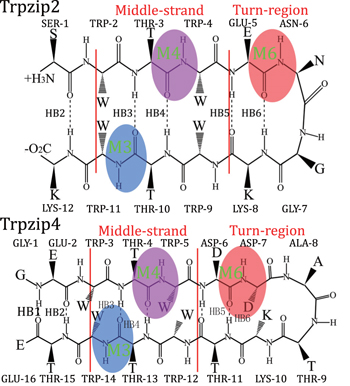
To further explore the capabilities of the isotopic Fourier transform IR (FTIR) and 2DIR melting spectra, we adapted this simulation protocol to identify the differences in peptide folding behaviors. Two hairpin polypeptides, Trpzip2 and Trpzip4, [ 81 ] with similar structures were compared (schematic structures for these two peptides are shown in Fig.
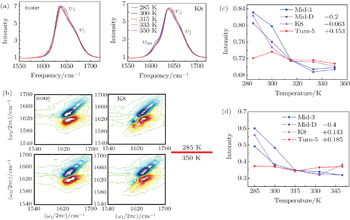
Simulated thermal unfolding FTIR and 2DIR spectra of Trpzip2 peptide, with the temperature range of 285–350 K, are presented in Fig.
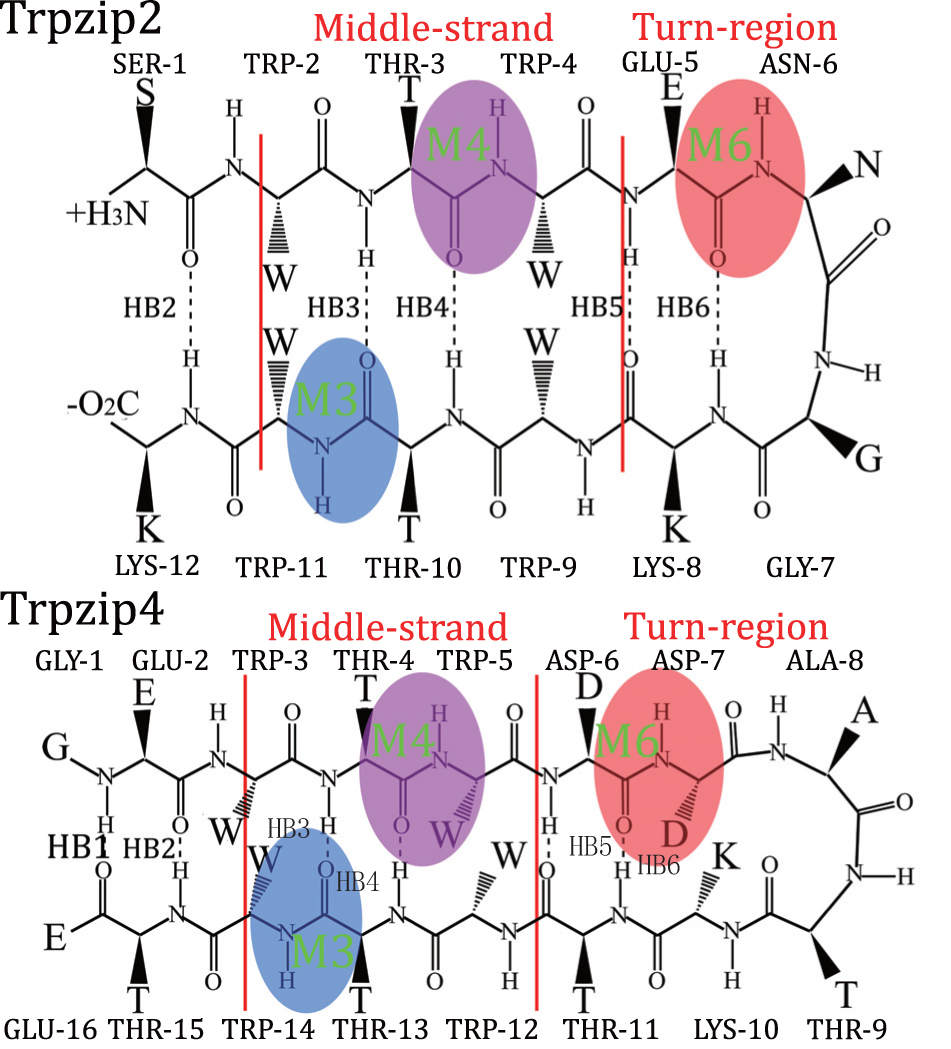 | Fig. 1. Structure of Trpzip2 (top row) with the isotope-edited sites TER- 1 (green), Mid-3 (blue), Mid-D (magenta), K-8 (yellow), and Turn-6 (red) highlighted, and Trpzip4 (bottom row), just the isotopologs Mid-3 (blue), Mid-D (magenta), and Turn-6 (red) in peptide were highlighted. The amide-I mode for these three isotope-edited sites in each polypeptides are also highlighted: M3, M4, and M6. The backbone hydrogen bonds HB2–HB6 are also labeled. |
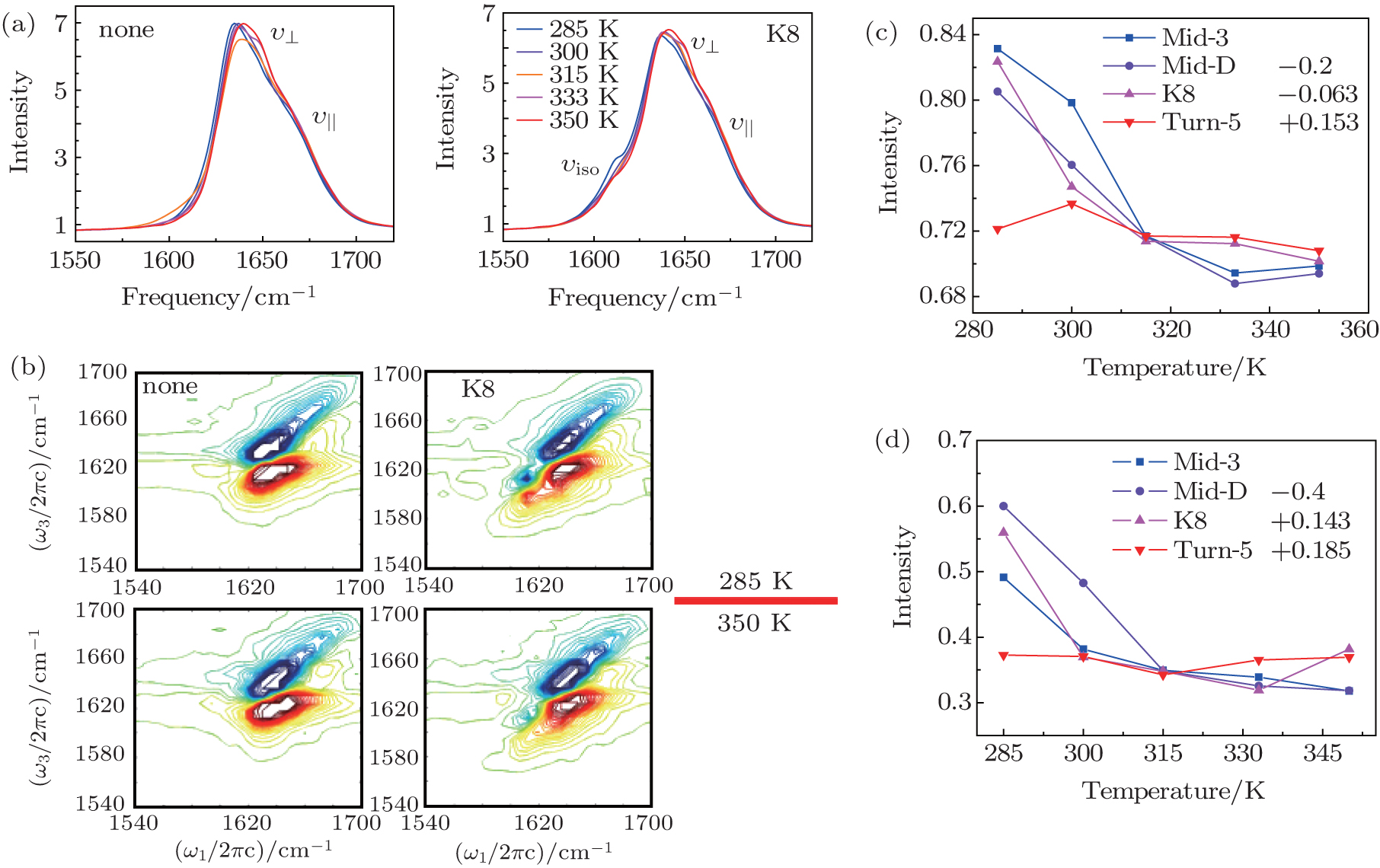 | Fig. 2. (a) Temperature-dependent simulated equilibrium FTIR spectra of isotope-unlabeled and isotopolog K8 in Trpzip2 taken from 285 to 350 K. Line color shows progression from blue to red. (b) The simulational 2DIR spectra of isotope-unlabeled and isotopolog K8 in Trpzip2 at 285 and 350 K. (c) Temperature-dependent intensities of the isotope-shifted peaks in the FTIR of different isotopologs. The curves of Mid-3, K8, and Turn-5 are shifted for better comparison. (d) Temperature-dependent intensities of the isotope-shifted peaks in the 2DIR spectra of different isotopologs. The curves of Mid-3, K8, and Turn-5 are shifted for better comparison. |
As temperature rises, each isotopolog has a significant blue shift for the domain peak υ ⊥ , while the shoulder υ || shows no frequency shift. The simulated 2DIR spectra for each isotopologue are demonstrated in Fig.
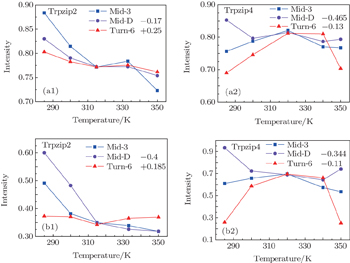
The temperature-dependent intensities of the isotope-labeled peaks are presented in Figs.
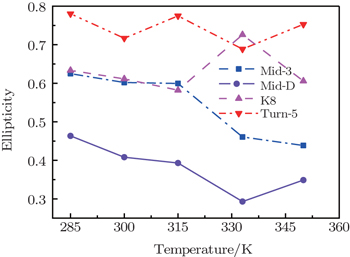
Value of ellipticity reflects the environment fluctuation around the amide-I mode. Ellipticity of a peak in the 2DIR spectra can be calculated as E = ( σ 2 − Γ 2 )/( σ 2 + Γ 2 ), [ 85 , 86 ] in which the diagonal line-width σ reflects inhomogeneous broadening, and the antidiagonal width Γ reflects the homogeneous broadening. Temperature-dependent ellipticities of the isotope-shifted peaks in the 2DIR spectra of different isotopologs were demonstrated in Fig.
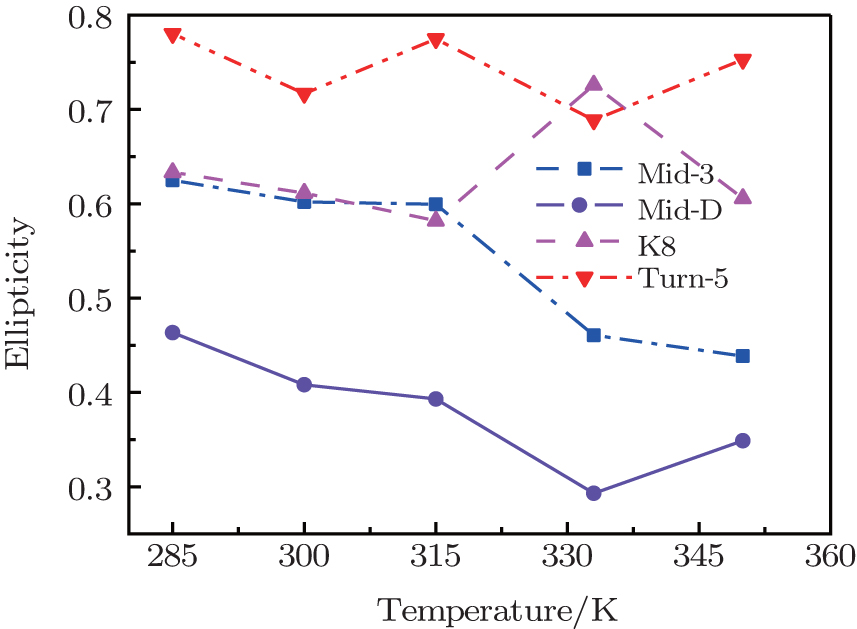 | Fig. 3. Temperature-dependent ellipticities of the isotope-shifted peaks υ iso in the 2DIR spectra of different isotopologs. |
Using the techniques developed, we further demonstrated that the different folding landscapes of two beta hairpin polypeptides with similar structures, Trpzip2 and Trpzip4, can be discriminated by the thermal unfolding vibrational spectra. Three C 13 isotope labeling schemes, as presented in Fig.
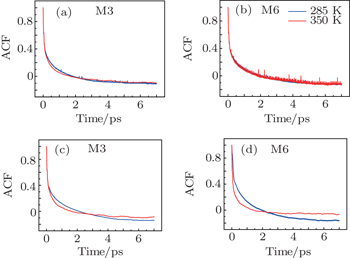
The time correlation function of the local amide-I modes M3 and M6 are demonstrated in Fig.
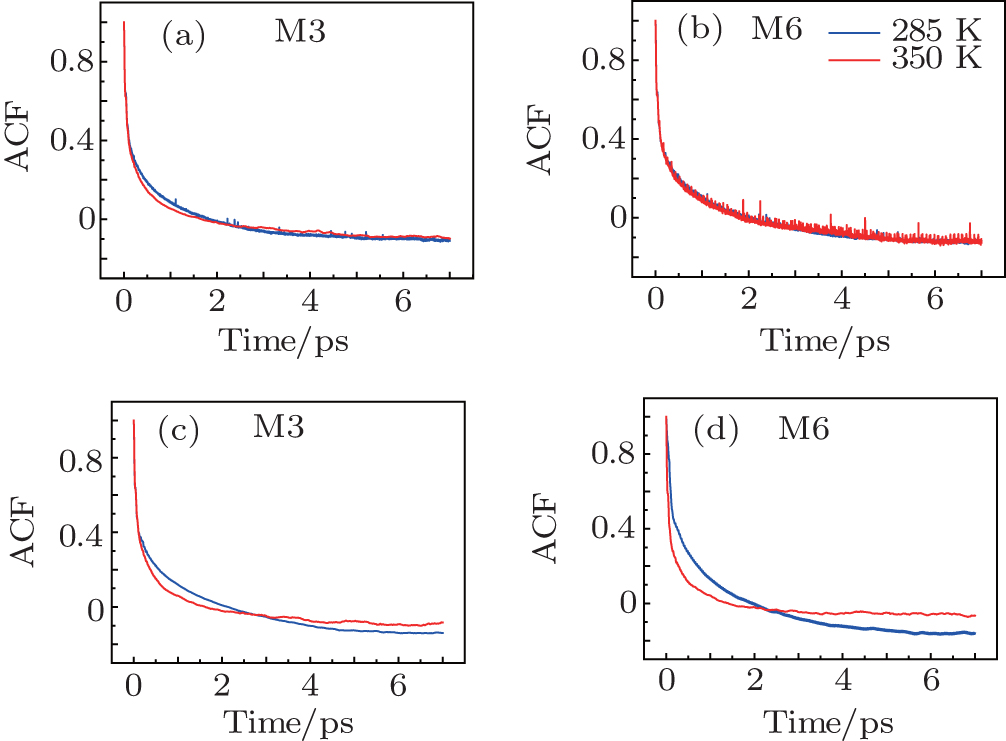 | Fig. 4. Frequency-time correlation functions of the localized amide-I modes ((a), (c) M3 and (b), (d) M6) for Trpzip2 (top row) and Trpzip4 (bottom row) at 285 and 350 K, respectively. |
The temperature-dependent intensities of the isotope-labeled peak or shoulder in the linear absorptions of all the three isotopologs are presented in Fig.
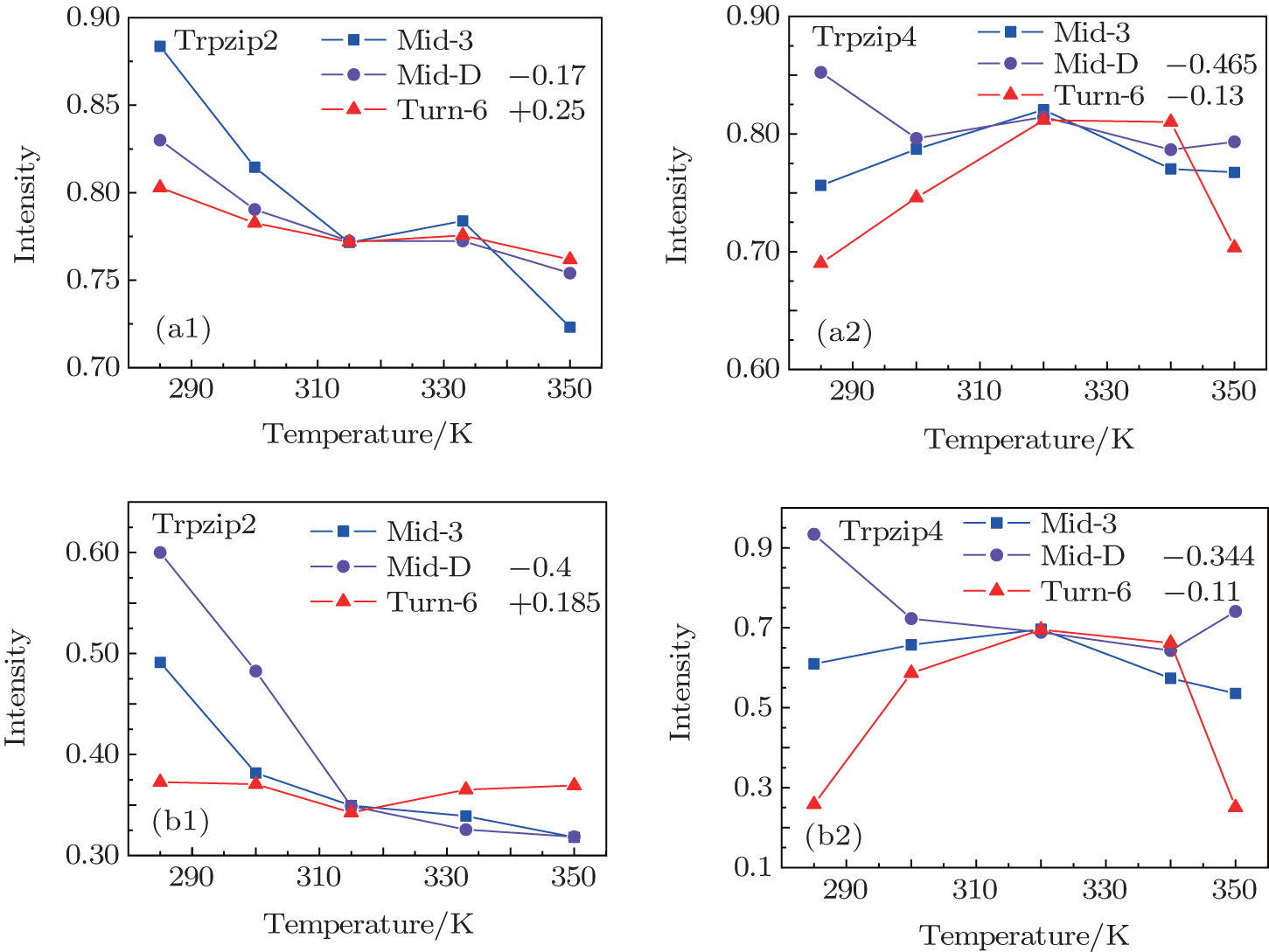 | Fig. 5. (a1), (a2) Temperature-dependent intensities of the isotope-shifted peaks υ iso in the linear absorption spectrum of different isotopologs. Both the curves of Mid-D and Turn-6 are shifted for better comparison. Temperature-dependent intensities of the isotope-shifted peaks υ iso in the 2DIR spectra for Trpzip2 (b1) and Trpzip4 (b2). The curves of Mid-D and Turn-6 are shifted for better comparison. |
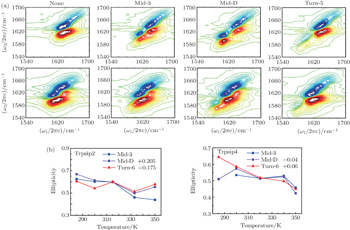
The 2DIR spectra of the unlabeled (none) and each isotopologue for both Trpzip2 and Trpzip4 polypeptides at 285 K are presented in Fig.
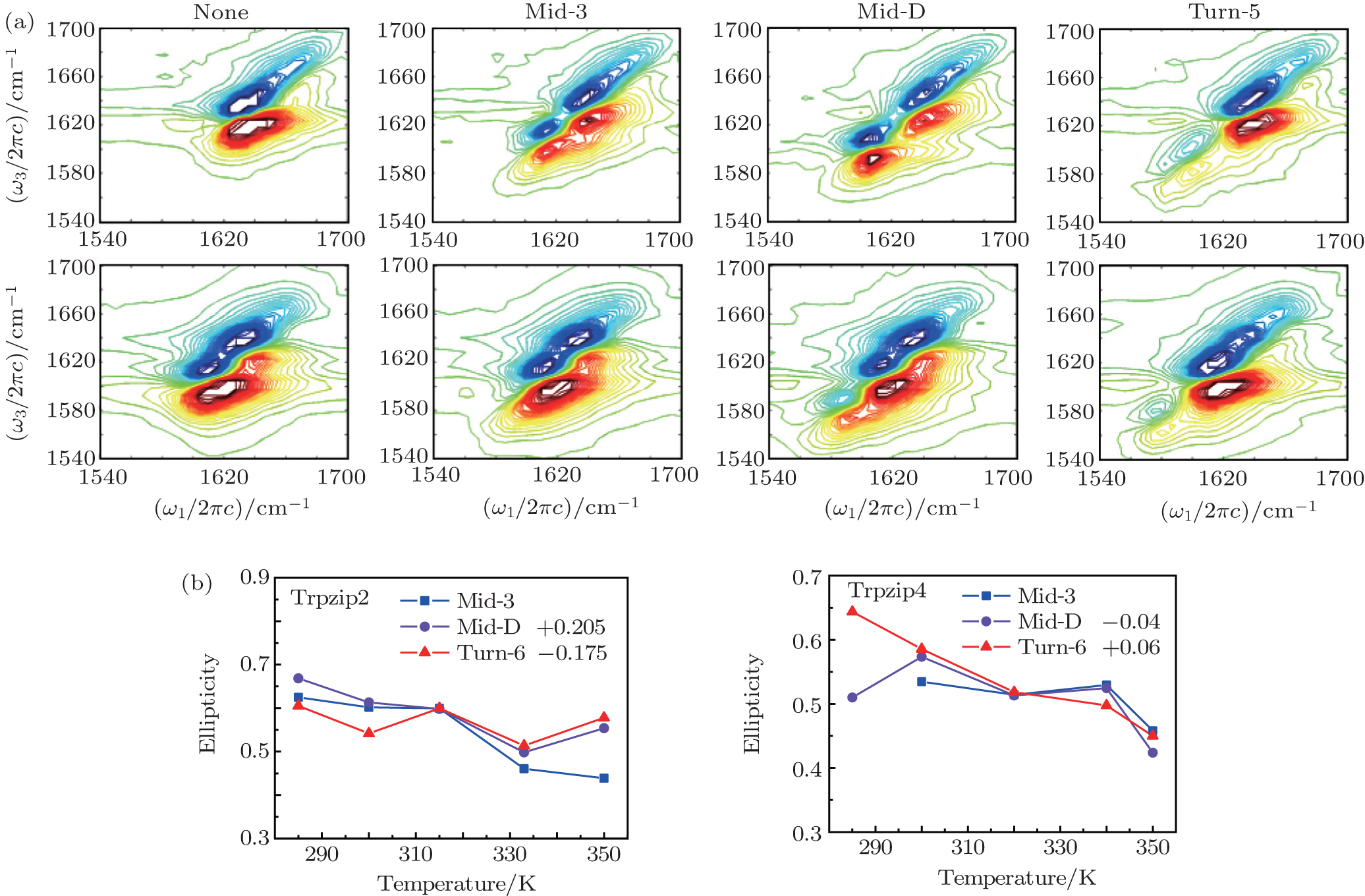 | Fig. 6. (a) 2DIR spectra of each isotopologue for both Trpzip2 (above) and Trpzip4 (below) are recorded at 285 K in simulation. (b) Temperature-dependent ellipticities of the isotope-shifted peaks in the 2DIR spectra of different isotopologs for Trpzip2 (left) and Trpzip4 (right). The curves of Mid-D and Turn-6 are shifted for better comparison. |
The isotope-labeled peak intensities of both FTIR and 2DIR demonstrate that different regions in Trpzip2 and Trpzip4 have different temperature dependencies. The isotope-shifted M6 peak in Turn-6 has the minimum intensity change in Trpzip2, and the maximum in Trpzip4. Other characteristics like the frequency distribution of vibrational modes, isotope-labeled peak shift, and full-width and half-maximum (FWHM), are not very sensitive to the local thermal stability. Furthermore, the intensity change of Turn-6 in Trpzip4 is non-monotonic. The intensity, therefore, is not aninformative marker for changesin local structure.
Temperature-dependent ellipticities of the isotope-labeled peaks of the 2DIR signals are presented in Fig.
The frequency distribution FWHM of the M3 and M6 modes are mostly constant as temperature increases. Therefore, the inhomogeneous broadenings of the isotope-labeled peaks are rather temperature-independent. The ellipticity is strongly correlated tovariations of the homogeneous broadenings, which are largely decided by the relaxation times of the frequency TCFs. The mid-strand segments in Trpzip2 peptide unfold first and thus the local environments around the M3 and M4 modes present much larger fluctuations compared to those around M6 in the turn-region. This difference in local structure stability induces a larger change in the frequency TCF relaxation time for the vibrational mode M3 than that for M6, and consequently a larger decrease in ellipticity. For Trpzip4, conversely, the local stability around M6 at the turn segment decreases more sharply than those around M3 and M4 at the mid-strand with increasing temperature, and eventually ellipticity presents a larger decrease for the Turn-6 isotopologue. Due to the sensitivity to homogeneous broadenings, the ellipticities of the isotope peaks in the 2DIR signals are more sensitive to these local stability differences than other spectral features like peak intensities; this suggests that the different folding landscapes for Trpzip2 and Trpzip4 polypeptides, the “hydrogen-bonded centric” and “hydrophobic-core centric” folding mechanisms, respectively.
We propose a cost-effective protocol to simulate FTIR and 2DIR spectra for polypeptides at a series of temperatures, based on comprehensive configuration samplings generated using implicit-solvent MD simulation combined with the ITS method, which provide an accurate thermal spectroscopic description consistent with experimental observables. Furthermore, to distinguish the differences inmelting spectra descriptions of peptide-folding behaviors, this protocol was adapted to simulate the thermal unfolding spectra for two β -hairpin peptides, Trpzip2 and Trpzip4, which were suggested to have different folding mechanisms. Comparison of the temperature-dependent signals for these two peptides, and the ellipticities of the isotope-labeled peaks were suggested to be more informative about the folding landscape differences, due to their sensitivity to homogeneous broadenings. Combined with the relevant experimental measurements, these modeling techniques can provide a better understanding of the peptide folding landscapes.
The amide-I vibrational band, although achieving great success in revealing secondary structure motifs of proteins, appears less sensitive to finer structural differences. For these “beats” differences, the amide III band, which mainly comprises backbone C–N stretching and N–H bending and is more delocalized than the amide I band, could be a potential solution. Moreover, the NEP method proposed here can work for any nonlinear optic process, including the surface-specified spectroscopy of sum frequency generation. Our next step will be to focus on using the NEP method together with the amide III band to investigate protein configuration dynamics in various chemical environments, which may help obtain a deeper understanding of fundamental life processes, like ion channels and photosynthesis. To further improve the current protocol, more close collaborations between the experimentalists and theorists are required.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 | |
| 52 | |
| 53 | |
| 54 | |
| 55 | |
| 56 | |
| 57 | |
| 58 | |
| 59 | |
| 60 | |
| 61 | |
| 62 | |
| 63 | |
| 64 | |
| 65 | |
| 66 | |
| 67 | |
| 68 | |
| 69 | |
| 70 | |
| 71 | |
| 72 | |
| 73 | |
| 74 | |
| 75 | |
| 76 | |
| 77 | |
| 78 | |
| 79 | |
| 80 | |
| 81 | |
| 82 | |
| 83 | |
| 84 | |
| 85 | |
| 86 |