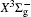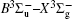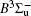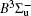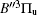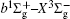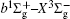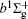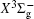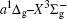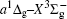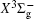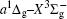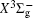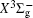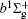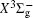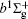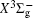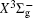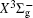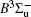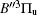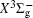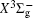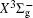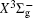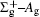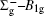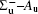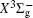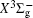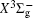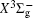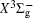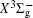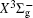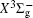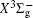Wei Changli, Zhang Xiaomei, Ding Dajun, Yan Bing. Accurate spectroscopic constants of the lowest two electronic states in S 2 molecule with explicitly correlated method. Chinese Physics B , 2016, 25(1): 013102
Permissions
Accurate spectroscopic constants of the lowest two electronic states in S 2 molecule with explicitly correlated method
Project supported by the National Natural Science Foundation of China (Grand No. 11574114) and the Natural Science Foundation of Jilin Province, China (Grand No. 20150101003JC).
Abstract
Abstract
A computational scheme for accurate spectroscopic constants was presented in this work and applied to the lowest two electronic states of sulfur dimer. A high-level ab initio calculation utilizing explicitly correlated multireference configuration interaction method (MRCI-F12) was performed to compute the potential energy curves (PECs) of the ground triplet and first excited singlet a 1 Δ g states of sulfur dimer with cc-pCV X Z-F12( X = T, Q) basis sets. The effects of Davidson modification, core–valence correlation correction, and scalar relativistic correction on the spectroscopic constants were examined. The vibration–rotation spectra of the two electronic states were provided. Our computational results show excellent agreement with existing available experimental values, and the errors of main spectroscopic constants are within 0.1% order of magnitude. The present computational scheme is cheap and accurate, which is expected for extensive investigations on the potential energy curves or surfaces of other molecular systems.
The homonuclear diatomic sulfur molecule (S 2 ) was observed in cometary atmosphere, [ 1 – 3 ] Jupiter’s atmosphere after the impact of the Shoemaker–Levy 9 comet, [ 4 ] and interstellar dense molecular clouds. [ 5 – 7 ] Moreover, S 2 has been found in various sulfur-containing compound plasmas and is regarded as an important intermediate in the combustion process containing sulfur elements. As the system is isovalent with the oxygen molecule, S 2 is closely related to the fields of photochemistry of Venus’s atmosphere [ 8 – 10 ] and circumstellar photochemistry. [ 11 ]
Over the past decades, the electronic structure and spectroscopic properties have attracted wide theoretical and experimental research interest, [ 7 , 12 – 21 ] in which amounts of accurate spectroscopic parameters of the S 2 dimer were systematically summarized by Huber and Herzberg [ 21 ] in 1979. Since the bands of the system in the UV region were first observed by Graham in 1910, [ 12 ] early observations focused on the spin-allowed transitions in S 2 , and many experimental studies (for reference, see [ 13 – 16 , 22 ] and references therein) focused on the predissociation of state, and perturbations between state and nearby electronic states like triplet and singlet 1 Π u states. Besides, the spin-forbidden, i.e., magnetic dipole and electric quadrupole transitions in O 2 and its isovalent systems were recorded (SO, [ 23 , 24 ] O 2 [ 25 – 28 ] ), and the spectroscopic constants and vibrational–rotational levels were experimentally determined. Similarly, as for the isovalent system, S 2 , Fink et al . [ 18 ] in 1986 recorded the emission spectrum of the system in near UV region, that is, 0–0, 0–1, 1–1 bands of the transition utilizing a Bomem interferometer with high resolution, while the accurate molecular constants of the and the states were deduced in the literature. In 2003, Setzer et al . [ 19 ] observed the 0–0 emission band of the magnetic dipole moment transitions for the sulfur dimer in near 4400 cm –1 infrared region, and the weak medium-resolution transition spectra of system of the sulfur molecule were observed using a Fourier-transition spectrometer, and the rotational constants of the a 1 Δ g and states were determined. At the same time, they evaluated the T 00 = 4394.25 ± 0.2 cm –1 of the 0–0 band of the system for the sulfur dimer. Furthermore, Green et al . [ 17 ] refined the spectroscopic constants of the vibrational energy levels ( ν ″ = 0–5, 7) for the ground state by analysis of the laser-induced fluorescence spectra of the molecule. Furthermore, the bond energy D 0 of the sulfur dimer was determined by Frederix et al. [ 7 ] in the photodissociation of the molecule for the first dissociation limit by velocity map imaging and the ionization energy value were obtained by Liao et al. [ 20 ] through the analysis of high-resolution photoionization spectra of the sulfur dimer in near infrared region of 600–1350 cm –1 using an oven-type supersonic beam method.
Very recently, motivated by singlet–triplet experimental observations in S 2 dimer, the spectroscopic properties of low-lying singlet electronic states attracted much theoretical interest. [ 29 , 30 ] The ab initio investigations on the low-lying states in S 2 have a long history. Swope et al. [ 31 ] first performed ab initio configuration interaction calculations on the ground and low-lying excited states. Then, Theodorakopoulos et al . [ 32 ] investigated the , a 1 Δ g , and states using large-scale configuration interaction method employing atomic orbital (AO) basis sets, and some spectroscopic constants for the three states were obtained in this literature. Subsequently, Hess and Buenker [ 33 ] performed the multireference single- and double-excitation configuration (MRDCI) wavefunctions and evaluated the spectroscopic constants values of ω e , R e for the , a 1 Δ g , and states. Later, Mclean et al. [ 34 ] undertook the self-consistent field (SCF) calculation for the ground state of second row diatomic molecules and cation molecules at the basis set limit for SCF and limited CI wave functions, and estimated the spectroscopic constants of the state. Moreover, Pradhan et al . [ 35 ] determined the potential energy curves (PECs) and the spectroscopic constants of the , , and states of sulfur dimer utilizing multireference configuration interaction (MRCI) calculations with correlation consistent basis sets. In 2000, Kiljunen et al . [ 36 ] deduced the spectroscopic parameters and the PECs of the , a 1 Δ g as well as eleven low-lying bound states for the sulfur dimer in solid argon through MRCI calculation with the correlation consistent cc-pCVQZ basis set. In 2004, Denis [ 37 ] estimated the bond length and harmonic constants of the ground state for the S X ( X : first- or second-row atom) diatomics. In 2010, Karton and Martin [ 38 ] evaluated some spectroscopic parameters ( ω e , ω e χ e , α e , and R e ) of the ground-state via performing the W4 and post-W4 theories. More recently, Xing et al . [ 39 ] calculated the PECs and the spectroscopic constants of some low-lying states including the and a 1 Δ g with conventional MRCI method and extrapolated to complete basis set (CBS).
In this work, we present a computational scheme to get accurate spectroscopic constants of a diatomic molecule, and choose the lowest two electronic states of S 2 as a case study. In the determination of the spectroscopic constants, the interpolation method is used to solve the radial Schrödinger equation. We performed the explicitly correlated multireference configuration interaction method for the lowest and a 1 Δ g states of the sulfur dimer with Davidson correction (+ Q), core–valence (CV) effect and scalar relativistic (SR) effect (mass–velocity term and Darwin term). Moreover, on the basis of the computed PECs of the two states, we computed the spectroscopic parameters, and the corrections effects on spectroscopic constants. Finally, based on the PECs of the and a 1 Δ g states of the sulfur dimer determined by the MRCI-F12 approach, the vibration-dependent rotational constant B ν , centrifugal distortion constant D ν , and vibrational energy levels G ν of the two states were also obtained by numerically solving the one-dimensional ro-vibrational Schrödinger equations.
2. Methods and computed details
In this work, a high level ab initio calculation for the sulfur dimer was performed by the MOLPRO-2012 program package. [ 40 ] The point group of the sulfur molecule is D ∞ h . However, owing to the limitation of the procedure, all of the calculations were carried out in the D 2 h subgroup of the D ∞ h point group. The correlating relationships of irreducible representations between D 2 h and D ∞ h point groups are , , , Δ u – B 1u + A u , Δ g – B 1g + A g , Π u – B 2u + B 3u , and Π g – B 3g + B 2g . The correlation consistent basis sets cc-pV X Z-F12(V X Z) and cc-pCV X Z-F12 [ 41 ] (CV X Z) ( X = T, Q) were employed to describe the sulfur atom, at a series of given molecular internuclear distances between 1.5–6.0 Å for PECs of the two electronic states for the sulfur dimer. We firstly carried out the Hartree–Fock self-consistent field method to obtain the starting molecular orbital (MO) wavefunctions. Subsequently, the PECs of the and a 1 Δ g states were yielded via the state-averaged complete active space self-consistent field (CASSCF) approach [ 42 , 43 ] by utilizing the reference wavefunctions from the Hartree–Fock method. In the CASSCF calculations, the active space consists of eight MOs: two A g , one B 3u , one B 2u , two B 1u , one B 2g , one B 3g symmetric MO, corresponding to the 3s3p atomic orbits of S, while ten MOs: three A g , one B 3u , one B 2u , three B 1u , one B 2g , one B 3g symmetric MOs closely correspond to the inner-shell 1s2s2p atomic orbits of S atom. Finally, we carried out the explicitly correlated multireference configuration interaction (MRCI-F12) calculations [ 44 ] for the sulfur molecule, in which the CASSCF wave functions were treated as the expanding referenced wave functions of the MRCI-F12 method. In addition, the size-consistency correction was taken into account through Davidson correction. [ 45 ] The core–valence correlation correction calculations [ 46 ] induced by the n = 2 shell of S atom were estimated by MRCI-F12 method and the 1s orbital of the S atom was excluded in the CV calculations. Moreover, we also introduced the scalar relativistic effect into our study utilizing second-order Douglas–Kroll [ 47 ] and Hess (DKH) [ 48 ] one-electron integrals with MRCI method in combination with uncontacted aug-cc-pVQZ basis set. [ 49 ] Finally, on the basis of the PECs plotted by different method, we obtained the spectroscopic parameters of the and a 1 Δ g states of the sulfur dimer by solving the nuclear Schrödinger equations utilizing numerical integration LEVEL procedure. [ 50 ]
3. Results and discussion
3.1. Spectroscopic constants
In our work, the high-level calculations on the ground have been carried out at different levels of theory. The PECs of the is calculated with the step lengths of 0.05 Å for R = 1.5–3.5 Å, 1.0 Å for R = 4.0–6.0 Å. The corresponding spectroscopic constants are determined by solving the corresponding nuclear Schrödinger equations and numerically tabulated in Table 1 , including the adiabatic excitation energy T e , vibrational constants ω e , and ω e χ e , equilibrium internuclear distance R e , rotational constant B e , and vibrational–rotational coupling constant α e , and dissociation energy D e . The related theoretical and the experimental results reported in the literature [ 7 , 21 , 22 , 34 – 39 ] are also listed in Table 1 for comparison.
Table 1.
Table 1.
Table 1.
Spectroscopic constants of state at different levels.
Spectroscopic constants of state at different levels.
.
The spectroscopic constants of the ground state are obtained from the MRCI-F12 calculations with and without the Davidson correction, SR effect, and CV effect employed triple and quadruple zeta basis sets. As shown in Table 1 , for the spectroscopic constants, both the values computed at MRCI-F12/VQZ level and those from VTZ basis set are very close to computed values extrapolated from complete basis set. Additionally, according to the present estimated values, we found the dependence on the basis set of the spectroscopic constants are not sensitive, the differences for the computed values at the MRCI-F12 level between VTZ and VQZ basis sets are merely less than 1 cm –1 for harmonic constant ω e , 0.1 cm –1 for ω e χ e , and on 10 –4 angstrom order. However, the basis sets effect on dissociation energy D e of the is relatively significant, the D e value improves ∼ 0.036 eV (∼ 8%) when VQZ basis set is adopted. However, though the basis set effect is not significant for the spectroscopic constants of the , slight improvements are still observed at larger basis set. Similarly, as displayed in Table 1 , the basis set dependence is not sensitive for the spectroscopic constants when considering Davidson correction, while computed values in the VQZ basis set are still closer to the accurate experimental values [ 7 , 21 ] than those in VTZ basis set. However, Davidson correction improves the dissociation energy significantly, the improvement reaches 0.11 eV both by VTZ and VQZ basis set; the correction makes the D e ’s deviation from experiment results [ 7 , 21 ] reduce to 10 –2 eV. Therefore, it indicates that the Davidson correction is important to overcome the size-extensivity problem for the MRCI-F12 approach as well as traditional MRCI method. Moreover, by comparing the computed results from CV X Z and those from V X Z ( X = T, Q) at the MRCI-F12 level, it is found that the CV correction shortens the equilibrium bond distance of the ground-state of S 2 , and reduce the error by ∼ 0.007 angstrom compared with the experimental value [ 21 ] of 1.8892 Å. At the same time, we found that the rotational constant value B e is improved with more accurate R e value. Compared with the experimental values, [ 21 ] the harmonic constant ω e improves by ∼ 4 cm –1 , while the ω e χ e value becomes slightly worse when introducing CV effect. It should be mentioned that the CV effect makes the dissociation energy value deviate from the experimental value, [ 21 ] only by considering the Davidson correction, the D e value becomes comparable with previous MRCI/CBS value, [ 35 ] and the deviation from experimental values [ 21 ] reduces to < 0.02 eV. Furthermore, we discuss the influence of the SR effect on the spectroscopic constants. According to Table 1 , the SR correction improves R e by 0.0001 angstrom, which is consistent with the previous computational result. [ 51 ] The SR effect on B e and α e is insignificant, and the SR correction improves ω e and ω e χ e by –1.87 and –0.01 cm –1 , respectively, but the SR correction significantly improves D e value [ 21 ] by –0.014 eV. For the dissociation energy value, debates still exist in experiments. [ 7 , 21 ] Hence more theoretical efforts are needed to explore the behaviors of PECs in the ground atomic asymptote, especially the spin-orbit interactions between ground state and excited states. In the present work, we follow the compiled experimental value [ 21 ] of D e . Finally, with inclusion of all corrections discussed above, we list the computed spectroscopic constants at MRCI-F12 + Q/CV X Z + SR ( X = T, Q) level. Generally, as exhibited in Table 1 , the MRCI-F12+Q/CVQZ+SR method give better dissociation energy D e and anharmonic constant α e , which are important to obtain high vibrational levels. Comparing with experimental results, [ 21 ] the deviations of our present computational strategy at MRCI-F12+Q/CVQZ+SR level are ∼ 0.001 angstrom (∼0.5%) for R e , < 2 cm –1 (0.3%) for harmonic frequency, and ∼ 0.003 eV (∼ 25 cm –1 , 0.07%) for D e , other spectroscopic constants are almost the same as the experimental values. [ 21 ] Moreover, the present computations are very cheap with ∼ 10 6 configuration state functions and could be performed on a personal computer.
With the same computational scheme, we perform MRCI-F12 computations to obtain PECs of a 1 Δ g state, and the fitted spectroscopic constants are listed in Table 2 . On the whole, from Table 2 , the computational results of a 1 Δ g state at the different levels also confirm the conclusions summarized above. The comparison between the computational results at MRCI-F12+Q/CVQZ+SR level and the experimental values [ 19 , 21 ] indicates that the errors of the R e and ω e are 0.0004 angstrom and less than 1 cm –1 , respectively. Our computed values of ω e χ e , B e , and α e are all well reproduced by the experiments. [ 19 , 21 ] To the best of our knowledge, no experimental value for dissociation energy of a 1 Δ g state is found. Our computed values are close to the previous theoretical ones. [ 36 – 39 ] For the transition energy T e , the early experiment reported an approximate value [ 21 ] of 4700 cm –1 , while the previous theoretical values [ 32 – 36 ] differ from each other. The recent experimentally determined T 00 value [ 19 ] is 4394 cm –1 . So according to T 00 value, [ 19 ] we estimated the T e of the a 1 Δ g state is 4405 cm –1 within conclusion of experimental zero-point energy. From Table 2 , our computed value T e are 4395 and 4381 cm –1 determined at the MRCI-F12 + Q/CVQZ + SR and MRCI-F12 + Q/CVQZ + SR levels, respectively, with deviations of less than 25 cm –1 (0.5%) from the deduced experimental value 4405 cm –1 .
Table 2.
Table 2.
Table 2.
Spectroscopic constants of a 1 Δ g state at different levels.
Estimated with experimental T e and zero-point energy (1/2 ω e – 1/4 ω e x e ).
Table 2.
Spectroscopic constants of a 1 Δ g state at different levels.
.
3.2. Rotation–vibration spectrum
On the basis of the PECs deduced from the MRCI-F12+Q/VQZ+CV+SR computations, we obtained vibrational information of the and a 1 Δ g states. In total 77 vibrational levels for the state and 71 vibrational levels for the a 1 Δ g state are found, and only the first 10 levels of the two states are listed in Tables 3 and 4 , respectively. As listed in Table 3 , only a few experimental values of vibrational energy levels are available, that is, v = 0–5 and v = 7 levels. For the v = 0–5 vibrational levels, the deviations between present work and experiment results [ 17 ] are in the range of 2–10 cm –1 ; for the v = 7 level, our computed value differs from the experimental one by 12 cm –1 , [ 17 ] while the relative errors for all available energy levels are in the range of 0.2%–0.3%, indicating the extensibility of present computational scheme. Since no experimental values for vibrational levels of a 1 Δ g state are found, we only list our calculated values. Our computed rotational constants for each given vibrational levels G v , B v , and D v , are also listed in Tables 3 and 4 for further comparisons with experiments.
Table 3.
Table 3.
Table 3.
Vibrational levels G ν , vibrational term values G ( v ) = G v – G 0 and rotational constants ( B ν , D ν ) of for the sulfur dimer deduced from MRCI-F12(+Q)/CVQZ+CV+SR method (all data are in units of cm –1 ).
Vibrational levels G ν , vibrational term values G ( v ) = G v – G 0 and rotational constants ( B ν , D ν ) of for the sulfur dimer deduced from MRCI-F12(+Q)/CVQZ+CV+SR method (all data are in units of cm –1 ).
.
Table 4.
Table 4.
Table 4.
Vibrational levels G ν , vibrational term values G ( v ) = G v – G 0 and rotational constants ( B ν , D ν ) of a 1 Δ g for the sulfur dimer deduced from MRCI-F12(+Q)/CVQZ+CV+SR method (all data are in unit of cm –1 ).
.
ν
J
G ν
B ν
D ν /10 –7
0
0
4732.0
0.29166
2.030
1
0
5428.6
0.28997
2.036
2
0
6119.1
0.28829
2.042
3
0
6803.4
0.28660
2.048
4
0
7481.7
0.28490
2.055
5
0
8153.8
0.28321
2.062
6
0
8819.7
0.28150
2.068
7
0
9479.5
0.27980
2.078
8
0
10133.1
0.27809
2.085
9
0
10780.6
0.27638
2.094
Table 4.
Vibrational levels G ν , vibrational term values G ( v ) = G v – G 0 and rotational constants ( B ν , D ν ) of a 1 Δ g for the sulfur dimer deduced from MRCI-F12(+Q)/CVQZ+CV+SR method (all data are in unit of cm –1 ).
.
4. Conclusions
In this paper, we present a cheap computational strategy to obtain accurate spectroscopic constants of the lowest two electronic states in S 2 with recently developed, explicitly correlated MRCI-F12 approach. The computed PECs of the and a 1 Δ g states at the MRCI-F12/CVQZ level result in accurate spectroscopic constants with an error of 0.1% order of magnitude with inclusion of Davidson correction, CV correlation, and SR effect. Subsequently, the vibration–rotation spectra of the two electronic states are yielded for further experimental comparisons. Some conclusions are summarized as follows.
The present computational strategy shows good extensibility, which is expected to be applied extensively to other diatomic systems.
Le Roy R J 2007 LEVEL 7.5: A Computer Program for Solving the Radial SchrÖdinger Equation for Bound and Quasibound Levels University of Waterloo Chemical Physics, Research Report CP-663
51
Peterson K A Lyons J R Francisco J S 2006 J. Chem. Phys. 125 084314
Accurate spectroscopic constants of the lowest two electronic states in S 2 molecule with explicitly correlated method
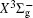
 ]
]