



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Soft x-ray spectroscopy for probing electronic and chemical states of battery materials
[Yang Wanli †,  , Qiao Ruimin ]
, Qiao Ruimin ]
, Qiao Ruimin ]


The formidable challenge of developing high-performance battery system stems from the complication of battery operations, both mechanically and electronically. In the electrodes and at the electrode–electrolyte interfaces, chemical reactions take place with evolving electron states. In addition to the extensive studies of material synthesis, electrochemical, structural, and mechanical properties, soft x-ray spectroscopy provides unique opportunities for revealing the critical electron states in batteries. This review discusses some of the recent soft x-ray spectroscopic results on battery binder, transition-metal based positive electrodes, and the solid-electrolyte-interphase. By virtue of soft x-ray’s sensitivity to electron states, the electronic property, the redox during electrochemical operations, and the chemical species of the interphases could be fingerprinted by soft x-ray spectroscopy. Understanding and innovating battery technologies need a multimodal approach, and soft x-ray spectroscopy is one of the incisive tools to probe the chemical and physical evolutions in batteries.
Lithium-ion batteries (LIBs), typically based on a transition-metal (TM) cathode, carbon anode, and carbonate electrolyte with additives, have conquered the electronic market requiring efficient energy storage, e.g., wireless communications and laptop computers, since 1991. [ 1 ] Now with the emerging clean energy applications, especially electric vehicles, the market of rechargeable LIBs is predicted to be exploded for ten times from 2010 to 2020. [ 2 ] However, the current battery technology is far below the standard to meet the formidable challenges for a low-cost, safe, high-capacity, and high-power energy storage solution.
The technological advance in battery technology is hindered by the complication of battery operation, which is typically beyond the thermodynamic stability of the electrolyte, and sometimes the electrodes. This instability requires a stable interphase layer on the carbon anode surface, now called solid-electrolyte-interphase (SEI), for LIBs. SEI was typically developed through decomposing carbonate based electrolyte with a number of electrolyte additives. Its functionality, design, and control remain the most critical R&D topics for LIB optimization and developments. [ 3 ] The recent developments of Si and Sn based high-capacity anodes encounters the same technical challenges on the SEI formation. For the cathodes, current commercial compounds are dominated by 3d TM based oxides and phosphates. Historically, the development of LiCoO 2 cathode was inspired by the consideration of the fundamental electronic structure of TM oxides. [ 4 ] Again, recent efforts on developing high-capacity and high-voltage cathode materials encounter technical challenges on how to stabilize the electrolyte and electrode materials at their interface. Such interface is under severe electrochemical instability due to the electron state configurations of high-voltage operations.
As a typical “electronic” device, an LIB operates with evolving electronic states in almost all the device components. The potential and kinetics of these electronic states define the transportation of the charges, i.e., electrons and Li-ions. More importantly, the relative configuration of the electron states fundamentally regulates the chemical activity at the surface and interfaces, thus determines the chemical reactions within the electrodes, as well as at the electrode–electrolyte interfaces. Such crucial chemical process could take place through either a direct redox reaction within the electrodes or electrolyte, or an indirect catalytic reaction at the interface.
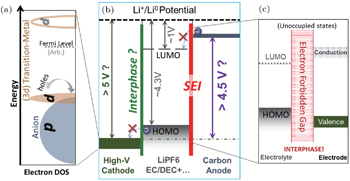
In order to elucidate the relationship between the fundamental electron states and the chemical activities in LIBs, we plot the electronic structure schematics of LIBs in Fig.
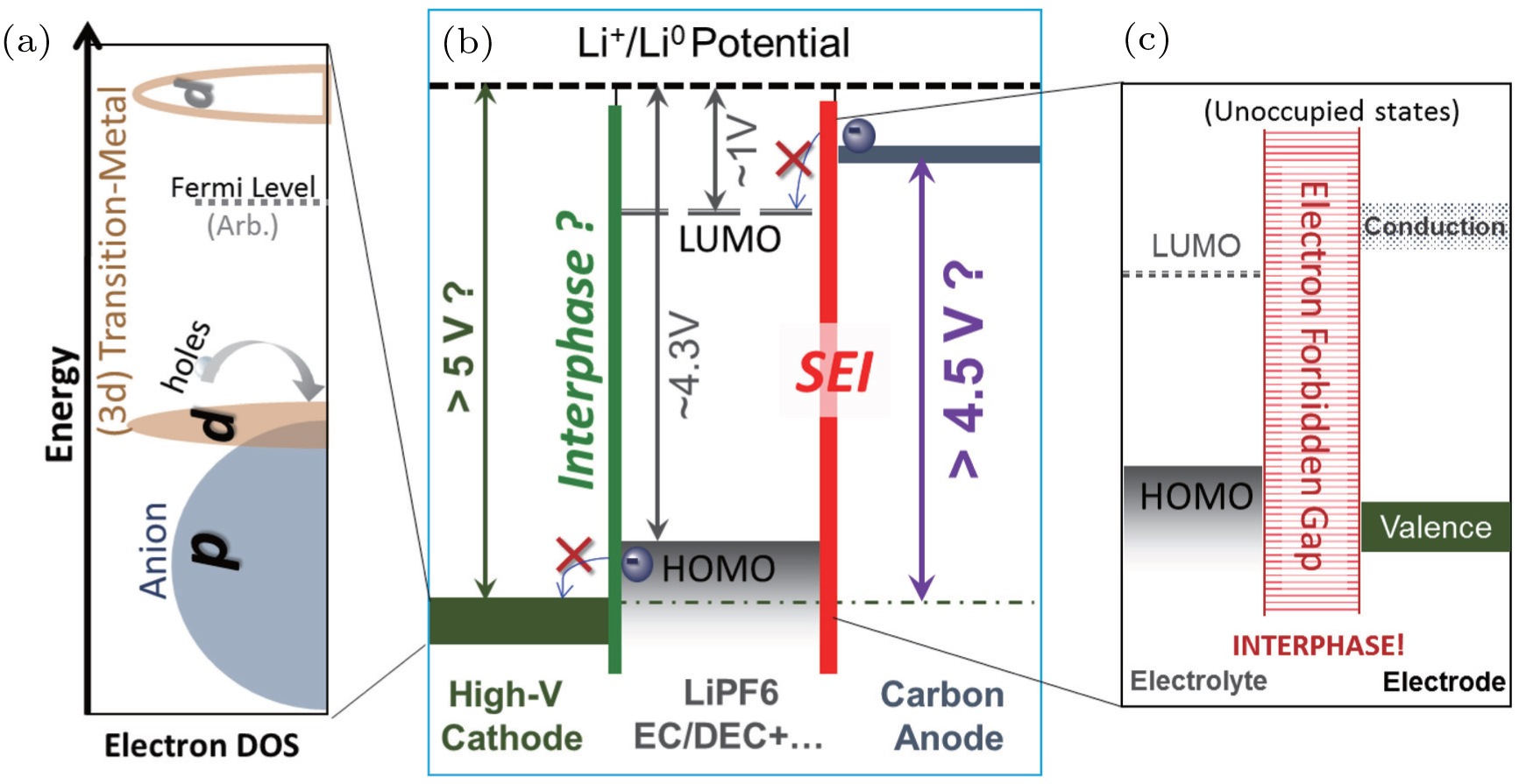 | Fig. 1. Schematic diagram of the critical electron states involved in an LIB. Panel (a) is the schematic of the relative energy positions of TM-3d and O-2p states in positive electrode materials. The dominating character of the highest energy occupied state determines whether a stable TM redox could be accessed during the charging (delithiation) process. This energy configuration also determines the catalytic property of the electrode material surfaces, which may destabilize the electrolyte material even within its electrochemical window shown in panel (b). Panel (b) shows the energy levels of a static open-circuit battery cell with carbonate based electrolyte without considering additives. Panel (c) displays the critical electron state configuration of a desired electrode–electrolyte interphase. This SEI diagram is general and plotted for both the negative and positive electrodes. Detecting, understanding, and controlling these key electron states remain formidable challenges and are directly related to the material and device optimization. |
An alternative way to achieve high power batteries is to form the passivating interphase layer on the surfaces of the electrodes. The most known is the SEI on the widely used carbon anodes, which is formed by decomposing carbonate based electrolyte with additives. [ 9 ] The interphase is typically formed by consuming the electrolyte components, but it does provide the kinetic stability to protect the electrolyte from further decomposition through the very large electron forbidden gap, as indicated in the right panel of Fig.
Besides the redox activity and charge transportation/forbidden at the interfaces in LIBs, the electron states in electrode materials also evolve vigorously throughout the electrochemical operations. Understanding the detailed electronic configuration beyond redox activity is particularly important for TM-based positive electrodes, as indicated in Fig.
Therefore, the electron state configuration of the unoccupied and occupied states, as well as the relative energy positions of the TM-3d and O-2p states, in LIB electrodes and electrolyte determines both the stability and performance of the battery cells. The interplay of these electron states provides the fundamental principles for understanding, designing, selecting and optimizing materials for the essential battery elements including electrodes, electrolyte, additives, and binders. These electron states in LIBs evolve with Li-ion diffusion at different electrochemical stages. Experimental probe of these evolving electron states in battery system is challenging but could be useful for obtaining the insights and guidelines for LIB material developments.
While soft x-ray spectroscopy detects both the occupied and unoccupied states of battery materials with inherent elemental and orbital sensitivity, [ 13 ] in this review, we focus on soft x-ray absorption spectroscopy (sXAS) that measures the unoccupied states with the existence of core-holes. We demonstrate that the information on electron states revealed by sXAS could be useful for understanding the chemical and physical process of LIB materials and interphases. The outline of this review is as follows. We first explain the basic principle of soft x-ray spectroscopy. A brief introduction of the state-of-the-art soft x-ray in-situ/operando instrumentation for studying electrochemical devices is included. We then showcase that the spectral features in sXAS represents the empty state that the electrons will occupy when the material is lithiated. Such information provides clear understanding and guidelines for optimizing the electronic properties of battery binder materials. Several examples on sXAS studies of TM-based positive electrodes are then discussed, where the redox could be quantitatively defined with elemental, orbital, and even site sensitivities. At the end, we show that although sXAS is a tool of detecting electron states, some chemical bond information could be derived because of the sensitivity of electronic structure to the chemical environment. We conclude that the critical chemical reactions in batteries, including both the redox evolution within battery components and the surface activities at the interfaces, could often be revealed by sXAS. sXAS is thus a powerful tool to reveal the chemical and physical evolution on a surface and/or interphase and complements many other techniques for measuring the structure and performance of battery materials.
Synchrotron-based core-level soft x-ray spectroscopy involves the excitation of core electrons, and the detection of emitted particles in the form of electrons and/or photons. Core-level soft x-ray spectroscopy typically includes x-ray photoelectron spectroscopy (XPS), soft x-ray absorption spectroscopy (sXAS), non-resonant soft x-ray emission spectroscopy (XES), and resonant inelastic x-ray scattering (RIXS). XES and RIXS have been employed to study battery materials, for detecting the occupied valence states, charge transfer, and electron spin states, etc. [ 14 – 19 ] Recent in-situ soft RIXS studies show that it could also be a powerful tool to detect the local chemical bonds of the solvation shell. [ 20 ] However, due to the limitation of the scope of this article, here we focus on the results from sXAS of LIB materials and interphases.
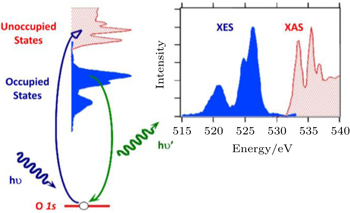
A simplified concept of sXAS and XES processes is shown in Fig.
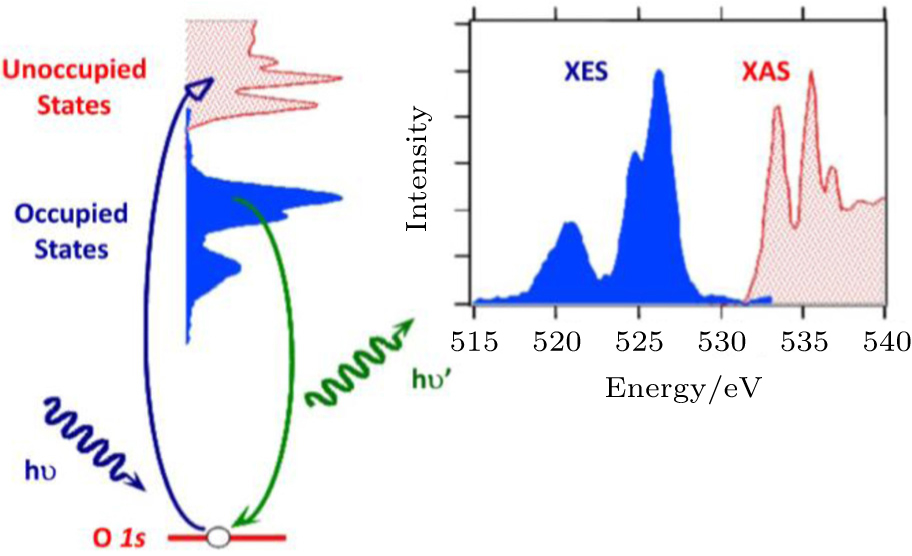 | Fig. 2. A schematic representation of soft x-ray absorption spectroscopy and emission spectroscopy processes. [ 21 ] sXAS and XES signals correspond to the unoccupied and occupied states, respectively. |
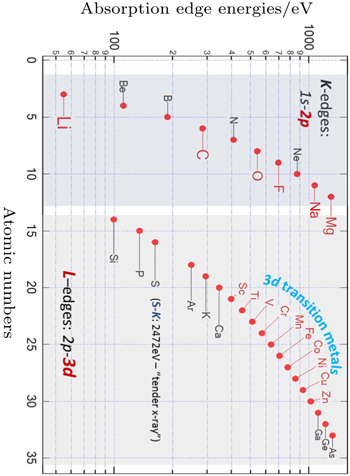
Modern synchrotron based sXAS has advantages over other techniques on probing the key electronic states in LIB materials. Firstly, sXAS is a core-level spectroscopy, the excitation of core electrons inherently provides the elemental sensitivity to core-level soft x-ray spectroscopies. Compared with hard x-ray (> 5 keV) techniques that are widely employed for diffraction and TM K -edge spectroscopic studies, the energy range of soft and tender x-ray (< 5 keV) covers the low- Z element K -edges, as well as the 3d transition metal L -edges (Fig.
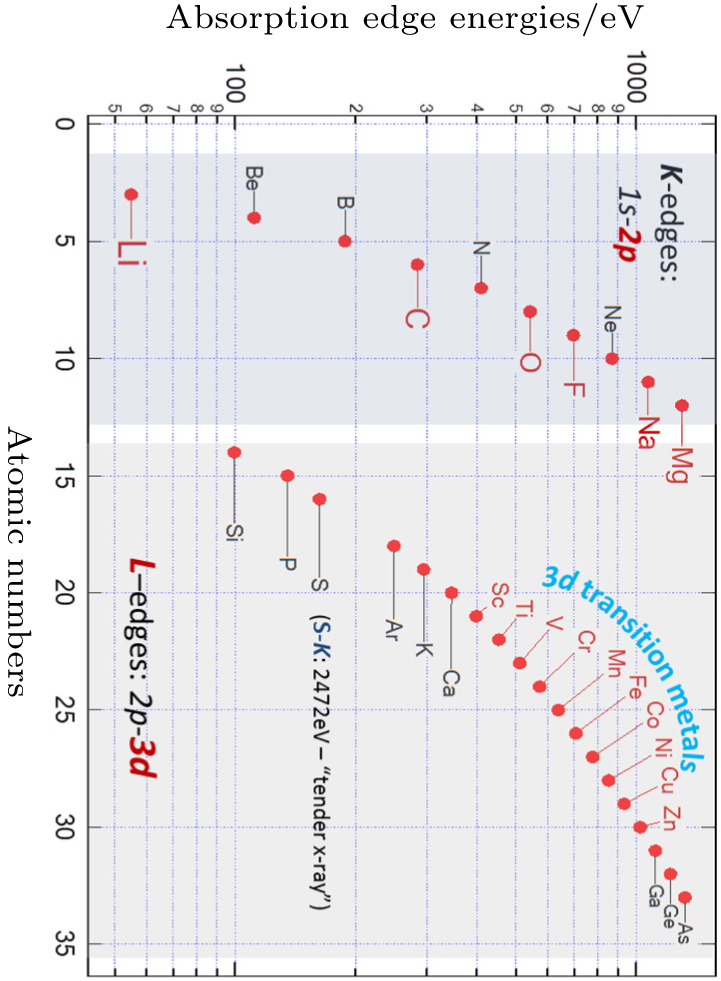 | Fig. 3. Coverage of elements that could be accessed by elemental-sensitive soft x-ray spectroscopy. K - and L -edge spectra correspond to the 2p and 3d electron states of the specific elements, respectively. [ 23 ] |
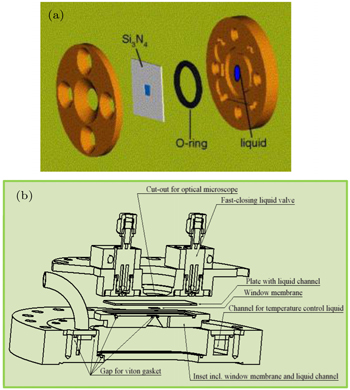
Secondly, sXAS could be performed in the PIPO mode with the probe depth of hundreds of nanometers in solid, depending on the energies and materials. This leads to the advantage on designing in-situ cells with sealed sample environment through soft x-ray membranes. Additionally, running spectroscopy in PIPO mode allows data collection of an electrochemical device under operando conditions with electric and magnetic fields. Various membrane windows with considerable soft x-ray transmittance have been employed for separating the liquid or gas environment from the UHV chamber. Two examples are shown in Fig.
sXAS is a direct probe of the excitations of core level electrons to the unoccupied states, i.e., the lowest-energy sXAS peak directly corresponds to the LUMO states of the interested material. Figure
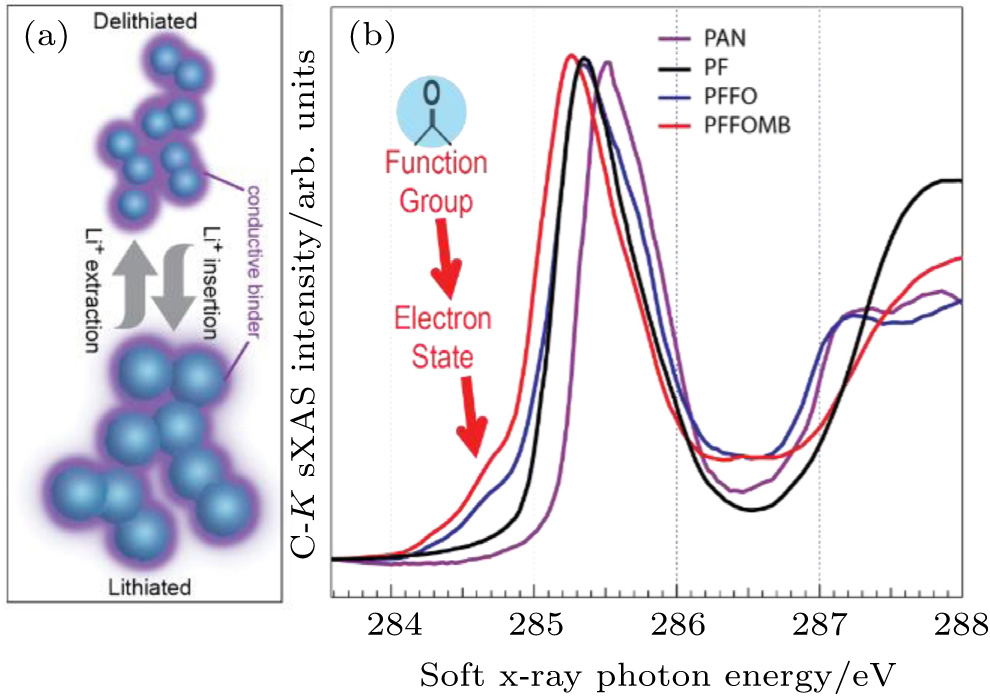 | Fig. 5. (a) Schematic of how a conductive binder could provide the dual functionality of both mechanical binder and conductive additives. (b) C- K XAS data collected on three selected PF type polymers, compared with the traditional p-type PAN. It is evident that the carbonyl groups in PFFO and PFFOMB generate a new LUMO state much lower than that of the PF (black) and PAN (purple). The XAS results are qualitatively consistent with ab-initio density functional theory (DFT) calculations, which confirms that this special low-energy electron state leads to an efficient electron doping to the backbone of the polymer, leading to greatly improved electric conductivity. [ 31 ] |
Binder system is a critical component in batteries, especially for high-capacity electrodes. Conventionally, nanoparticle active materials require high volume of conductive additive, such as acetylene black, to ensure high inter-particle electric conductivity. The high volume of additive reduces both volumetric and gravimetric lithium-ion storage capacity. In addition, because the conductive additive has no mechanical binding force, it turns to be pushed away from the high-capacity active materials by the severe volume expansion, leading to broken electric connections after extended cycles. [ 26 – 28 ] Therefore, an ideal battery binder should provide inexhaustible tolerance of the large volume change of high-capacity materials during battery operation. [ 29 , 30 ]
The sXAS observation of the special LUMO state in the binder polymers with carbonyl group indicates that the electrons introduced through the lithiation process will occupy this low-energy state. This is consistent with ab-initio density functional theory (DFT) calculations, which predicts an efficient charge transfer if the material is employed in LIB chemistry. Furthermore, the calculation shows that the spatial distribution of this low-energy electron state leads to efficient doping of electrons into the backbone of the PF polymers. [ 31 ] Such information strongly suggests that the special electron state observed in sXAS will result in an in-situ electron doping and will improve the electric conductivity if the material is used in a reducing (lithiation) environment. This clear understanding and further impedance tests provide a powerful guideline for further improving the materials. A new binder system is developed by optimizing the electronic and mechanical properties separately. [ 29 ] When we test the developed binder polymer with Si anodes, which suffer the most severe volume change problem, a composite anode without any conductive additive is demonstrated with a decent cycling stability.
Cathode materials sit in the heart of modern battery researches, partially because there is still no perfect candidate that could maintain high-power and stable cycling with capacity comparable to that of the anodes. [ 32 , 33 ] Although novel cathode systems such as conversion-type and organic cathodes are under scrutiny, the TM-based intercalation-type cathodes remain the most promising technology in the short/mid-term.
For TM-3d states, the L -edge sXAS is arguably the most sensitive probe through the dipole allowed 2p–3d transitions. Such sensitivity of sXAS provides the unique opportunity to detect abundant information of the TM-3d states in the oxidation (valence) states, ligand field, spin and orbital properties. [ 13 , 34 – 36 ] In general, the TM spectra can be divided into two regions, L 3 -edge at lower photon energy and L 2 -edge at higher energy, due to the 2p core hole spin-orbital splitting. The L 2 -edge display much broader absorption features than L 3 -edge due to the shorter lifetime of the 2p 1/2 core hole as a consequence of Coster–Kronig auger decay. [ 37 ] Therefore, the spectral analysis typically focuses on the L 3 -edge absorption fine features. Below, we discuss some of the recent sXAS results collected on the Fe and Ni L -edges of a series of battery cathodes. [ 34 , 36 , 38 ] Note that sXAS is applicable for a direct detection of 3d states for all TM- L and 2p states of O- K edges in battery cathodes. [ 35 , 39 – 42 ]
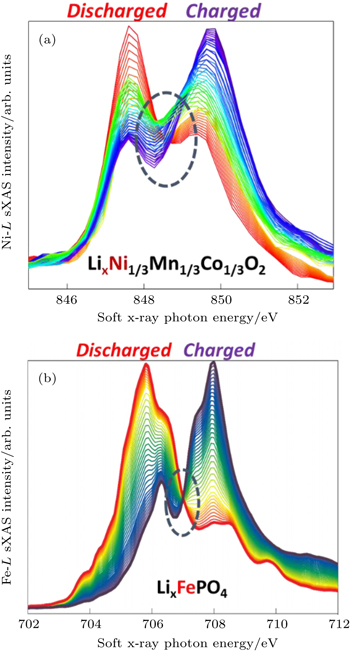
The sensitivity of soft x-ray photons to the TM-3d states is evidently testified by the data in Fig.
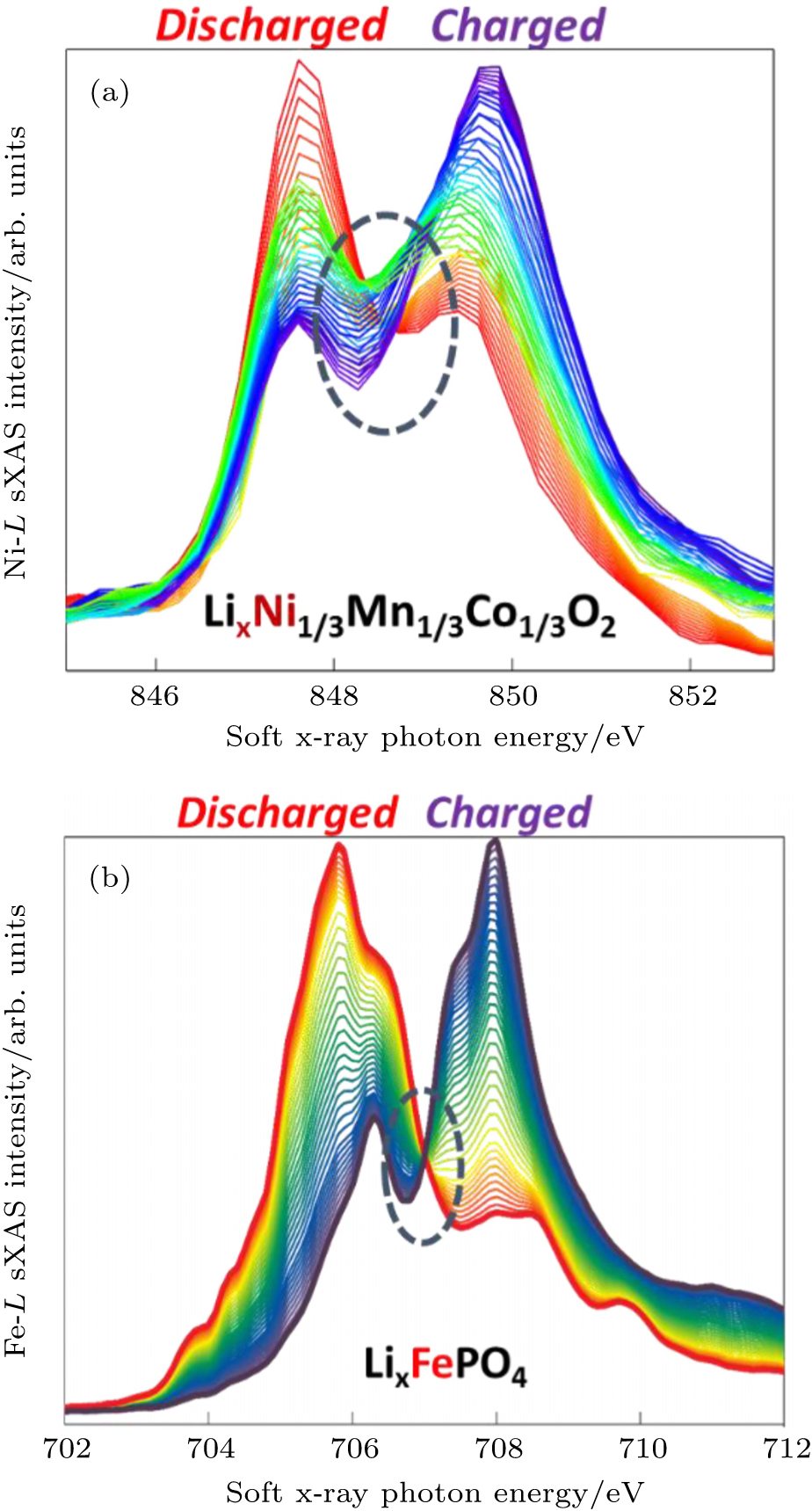 | Fig. 6. Ni- L and Fe- L edge sXAS spectra collected on Li x Ni 1/3 Mn 1/3 Co 1/3 O 2 (a) and Li x FePO 4 (b) electrodes under in-situ and ex-situ conditions, respectively. The red (dark purple) spectra correspond to the electrodes at fully discharged (charged) states. The evolution of the spectra represents the changes of the TM-3d states at different electrochemical stages. The well-defined isosbestic point of the Li x FePO 4 indicates a two-phase transformation. |
The sXAS of TM- L edges could benefit battery researches in two different aspects. Firstly, because of the high sensitivity of TM- L to the redox states of TM (Fig.
Secondly, because sXAS results correspond to the partial density of state (pDOS) of electrons with the existence of core holes. The TM L -edge data could serve as a bridge between the materials and theoretical calculations. Because of the localization character of the 3d states and the strong overlap of core and valence wave functions, the so-called multiplet effect, ligand-field multiplet theory could often be successfully employed for simulating TM- L sXAS results. [ 22 ] Such calculation delivers detailed information on the TM-3d electronic structure. For example, a combined theoretical and spectroscopic work has recently clarified the single electron charge transfer mechanism for a Ni-based positive electrode material. [ 44 ] In multiplet calculation, the molecular environment and local symmetry are described using a ligand-field potential. The multiplet calculations, by comparison with experimental data, will be able to deliver a complete set of parameters that defines the evolution of electronic structure affected by the local lattice distortion. [ 34 ]
A combined sXAS and multiplet calculation of iron hexacyanoferrate (HCF) is shown in Fig.
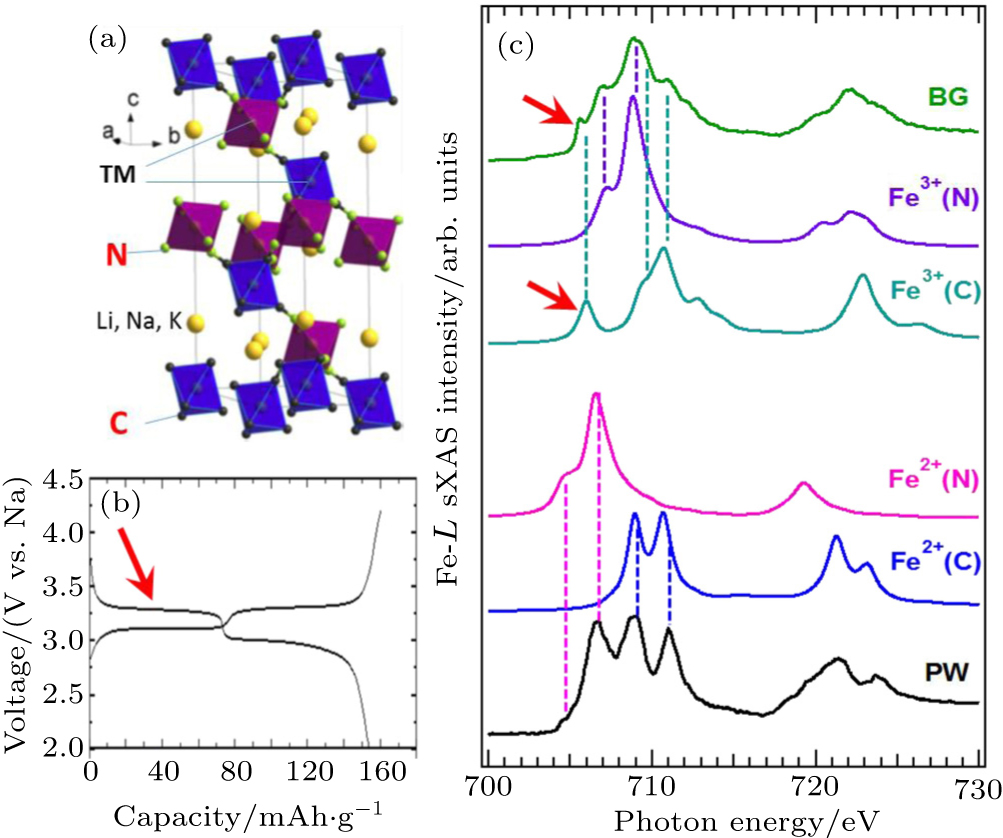 | Fig. 7. Experimental Fe L -edge sXAS spectra of Prussian White (PW, Na 2 Fe II [Fe II (CN) 6 ]) and Berlin Green (BG, Fe III [Fe III (CN) 6 ]). Panel (a) shows the lattice structure of the iron HCF materials. Panel (b) displays the two electrochemical potential plateau corresponding two redox reactions. (c) The resolved experimental features could be clearly assigned to Fe atoms at different sites with the specified formal valences. The lithiation (discharge) process introduces electrons into the lowest energy electron state in the BG Fe III spectrum (red arrow), which corresponds to the Fe III at the carbon coordinated site. This combined spectroscopic and theoretical study clarifies the mechanism of the high (low) electrochemical plateau from the low (high) spin Fe at the C (N) coordinated site. [ 36 ] |
The multiplet calculations accurately modeled the sXAS results of both the PW and BG (Fig.
The different spin states of Fe at different sites determine two different redox potentials as shown in the electrochemical tests (Fig.
While most TM based compounds are considered positive electrodes in batteries, Ti-based electrodes display potentials close to Li/Li + and are considered promisinganodes free of the SEI issues. [ 6 ] Furthermore, the Ti,Mn-based electrode is recently found to be a candidate of Na-ion battery anode. [ 39 ] Although sXAS studies on these TM based anode materials are very limited, [ 46 , 47 ] we note that the methodology of employing sXAS for revealing the redox and charge diffusion could be directly extended to the study of the anode materials. Additionally, detecting the oxidation states of TMs is not limited to a particular component in batteries. For example, sXAS Mn L -edge data clearly show that the Mn deposition in the anode SEI is di-valent. [ 48 ]
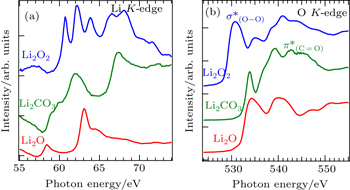
While sXAS corresponds to the electron pDOS of the interested materials, it could also serve as a chemical bond probe because different chemical bonds often lead to distinct electron states. Figure
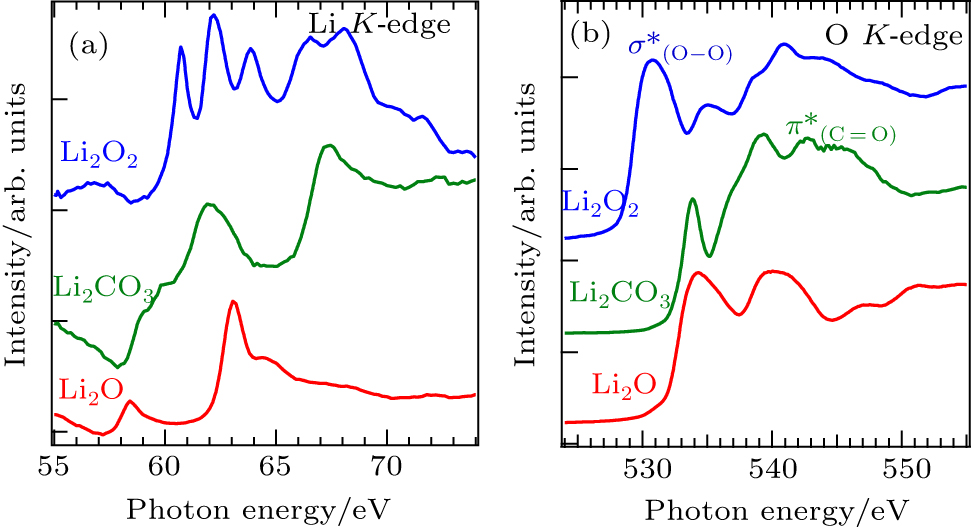 | Fig. 8. Li K -edge (a) and O K -edge (b) XAS spectra of Li 2 O 2 , Li 2 CO 3 , and Li 2 O. The different sXAS features are due to the distinct electron states from the particular chemical bonds. [ 52 ] |
For the O K -edge sXAS, the O 1s core hole is now well-screened compared with the shallow Li 1s core hole. Thus O- K spectra are less affected by excitonic effect and better reflect the partial density of states of unoccupied O 2p orbitals. All three lithium compounds display rather different absorption spectra due to variations in the local oxygen environment, and in particular the sharp features from the O–O (in Li 2 O 2 ) and C=O bond (in Li 2 CO 3 ). Note that the leading edge of Li 2 O 2 XAS spectrum is about 3.5 eV lower than that of Li 2 CO 3 and Li 2 O, suggesting that the conduction band minimum of Li 2 O 2 is lower than that of Li 2 CO 3 and Li 2 O due to its O–O bond. Therefore, sXAS of the low- Z element K -edges could be utilized for fingerprinting some of the chemical species involved in battery operations. [ 53 ]
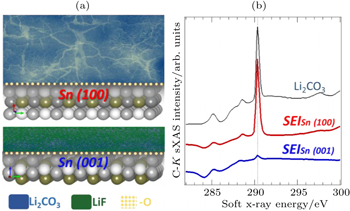
Figure
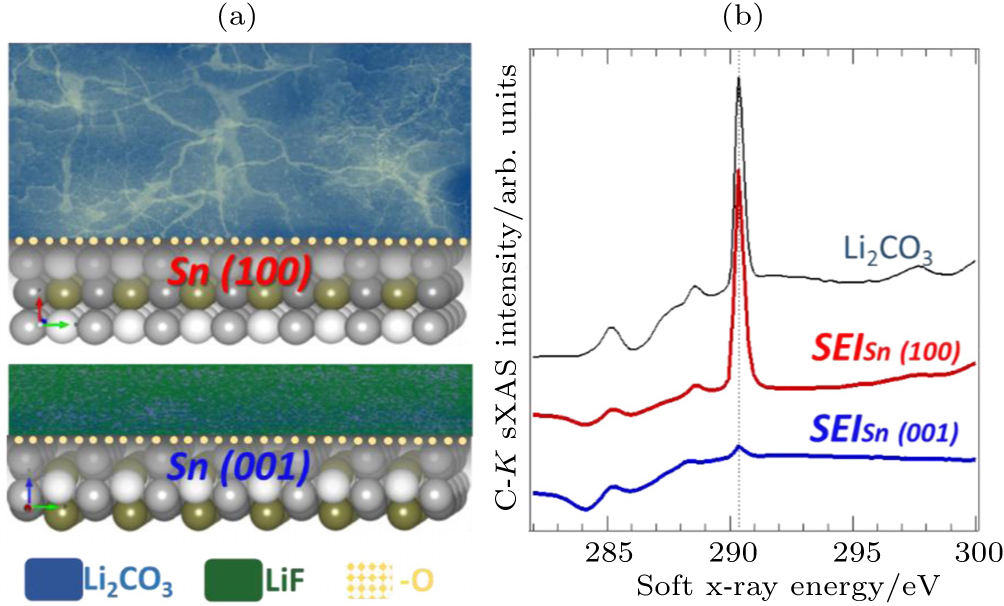 | Fig. 9. (a) Distinct SEIs formed on single crystal Sn electrodes with different surface orientation. (b) sXAS of C- K edges of the SEIs formed on Sn electrodes with two different surface orientations. sXAS of Li 2 CO 3 reference is plotted on top for comparison. The distinct SEIs are clearly revealed by the sXAS results of the relevant low- Z element K -edges, including the C- K , O- K , and F- K . [ 54 ] |
Due to various technical issues with carbon anodes, e.g., Li dendrite formation and SEI stability, developing better anode and electrolyte system is another critical challenge for batteries. We would like to emphasize that, developing advanced anode systems, e.g., Si and Sn, are always associated with the electrolyte optimization. The capability of sXAS to probe the surface properties with different probing depths makes it an advanced tool to study both the Si and Sn- based materials [ 54 , 56 , 57 ] and the interphases. Moreover, it has been recently recognized that the cathode SEIs also play important roles in the battery performance, especially for high voltage cathode materials, like LiNi 0.5 Mn 1.5 O 4 . [ 40 , 58 , 59 ] Despite of its importance, little is understood about the SEI formation, functionality and control. sXAS is inherentlya probe of the local chemical bonds and electronic states of surfaces and interfaces. Its surface sensitivity provides complementary, and often critical, information of the material surface in batteries. [ 47 ] Soft x-ray techniques have recently attracted much attention as a powerful tool to reveal the physical and chemical properties of the electrodes, electrolyte and interphases in batteries. Advanced instrumentation and methodology based on soft x-rays are still under developments for providing more and more specific and critical information for battery optimizations.
This review discusses the application of soft x-ray spectroscopy for studying various battery materials, including the electrodes and SEIs. We first introduce the basic principle of soft x-ray spectroscopy and then focus on sXAS studies of battery materials. Because sXAS detects the unoccupied electron states in the vicinity of the Fermi level, the sXAS spectral features correspond directly to the states that electrons will occupy during the lithiation process. We show that a special electron state revealed by sXAS is responsible to the superior electric property of a conductive battery binder. We also show that sXAS is an inherent elemental and orbital sensitive probe of the TM-3d and O-2p redox in positive electrodes. This capability makes sXAS arguably the best tool to clarify the cycling mechanism of TM based positive electrodes. In particular, directly fingerprinting the TM-3d states provides unique opportunities for detecting the charge diffusion process, sometimes with a microscopic picture under in-situ/operando conditions. [ 60 ] Additionally, because TM-3d states are strongly localized, atomic multiplet calculation could be employed to simulate TM- L sXAS spectra. This combination is powerful to reveal much information beyond the valence changes, e.g., spin states, hybridization, and crystal field with site sensitivity.
One must note that both the advantage and disadvantage of soft x-ray spectroscopy relies on its sensitivity to the electron states and to the surface with about 10 nm and 100 nm probe depth. Therefore, soft x-ray techniques are suitable for studying the interfaces and interphases in batteries. Some of the sXAS studies deliver the chemical reaction information, which sheds new lights on understanding the critical interface phenomena in batteries.
Battery electrolyte with additives, although critical for high energy-density batteries, is not discussed in this work. However, with the recent progress on in-situ soft x-ray experiments and with the inherent surface sensitivity of soft x-ray techniques, both the bulk liquid and solid–liquid interfaces could be studied now under operando conditions with various soft x-ray spectroscopy, including the sXAS, XES, RIXS, and ambient-pressure XPS. [ 20 , 25 , 43 ] While most electrolyte developments are trial-and-error based and strongly depend on the experience and subjective speculations of elite material scientists, we hope the utilization of soft x-ray techniques could open up new opportunities for both understanding and developing electrolyte materials for batteries.
At this time, a low-cost, safe and high-performance energy storage solution is still a daunting task for the scientific and industrial communities. A multimodal approach, including hard x-ray and soft x-ray techniques, imaging and spectroscopy tools, ex-situ and in-situ conditions, are needed to truly understand the complex electrochemical process in batteries. Soft x-ray spectroscopy, as a surface sensitive probe of electron states, has been constantly optimized to contribute to the field of batteries with Li-ions and beyond.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 | |
| 52 | |
| 53 | |
| 54 | |
| 55 | |
| 56 | |
| 57 | |
| 58 | |
| 59 | |
| 60 |