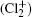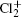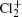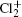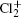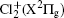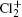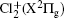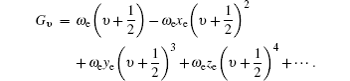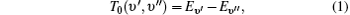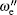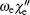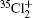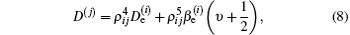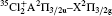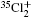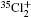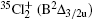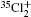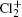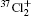Wu Ling, You Su-Ping, Shao Xu-Ping, Chen Gang-Jin, Ding Ning, Wang You-Mei, Yang Xiao-Hua†. Study on the A2Π3/2u, B2Δ3/2u, and X2Π3/2g states of including its isotopologues . Chinese Physics B, 24(8): 083301
Permissions
Study on the A2Π3/2u, B2Δ3/2u, and X2Π3/2g states of including its isotopologues
Wu Linga), You Su-Pinga), Shao Xu-Pingb), Chen Gang-Jina), Ding Ninga), Wang You-Meia), Yang Xiao-Hua†b)
School of Science, Hangzhou Dianzi University, Hangzhou 310018, China
School of Science, Nantong University, Nantong 226019, China
*Project supported by the Natural Science Foundation of Zhejiang Province, China (Grant Nos. Y6110524 and Y1111085), the Scientific Research Foundation of the Department of Education of Zhejiang Province, China (Grant No. Y201430970), the National Nature Science Foundation of China (Grant No. 11247007), and the QingLan Project of Jiangsu Province, China.
Abstract
Adopting the experimentally available vibrational constants in a recent analysis of the strong perturbation between the A2Π3/2u and B2Δ3/2u states of in the A–X band system [Gharaibeh et al. 2012 J. Chem. Phys.137 194317], an unambiguous vibrational assignment of the bands reported previously is carried out. The equilibrium rotational constants Be and αe of the X2Π3/2g and A2Π3/2u states for and35Cl37Cl+ and those of the B2Δ3/2u state for are obtained by fitting the experimental values of B υ. In addition, the values of Be and αe of these three states for the minor isotopologues35Cl37Cl+ and are predicted by employing the isotopic effect. The values of equilibrium internuclear distance Re of the three states for the three isotopologues are calculated as well.
The highly perturbed A2Π u– X2Π g system of the chlorine cation has been investigated for nearly one century, however, knowledge about the structure of the electronic states, A2Π u and X2Π g, is still limited. This system has two components: A2Π 1/2– X2Π 1/2 and A2Π 3/2– X2Π 3/2. Initial spectroscopic data came from a series of dispersed emission studies.[1– 5] It is difficult to assign the absolute vibrational quantum numbers to the observed bands because of the irregular vibrational internals and non-systematic chlorine isotopic effects resulting from the severely perturbed excited electronic state A2Π u. In addition, since the spin– orbit splitting for the X2Π g state is found to be very close to the vibrational energy spacing, the spin– orbit resolved vibrational bands have been analyzed ambiguously. In 1966, Huberman[6] studied the emission spectrum and made the most comprehensive rotational assignment of the observed. In 1989, Tuckett and Peyerimhoff[7] observed the emission spectrum in the supersonic expansion, which allowed them to make a fuller analysis due to the cold Doppler-free spectrum. One hundred vibronic bands were fitted into two Deslandres tables (one for A2Π 1/2– X2Π 1/2, and the other for A2Π 3/2– X2Π 3/2). Still, they could not assign vibrational quantum numbers to the Deslandres tables. Ab initio calculations show that A2Π u is crossed near its origin by some states arising from , i.e. and 2Δ u.
In 1989, the Λ -doublets was first observed in a jet-cooled A– X emission spectrum by Choi and Hardwick, [8] and four bands were unambiguously assigned as A2Π 1/2– X2Π 1/2. Later in 1991, they[9] reported more bands of the Ω = 3/2 component. Among those, 10 bands were assigned to the A2Π 3/2– X2Π 3/2 system, which had been rotationally analyzed by Huberman earlier. These measurements improved the accuracy of the rotational constants for this system. Additionally, five bands were assigned to a new system of B2Δ u3/2– X2Π g3/2, and they, for the first time, rotationally analyzed the bands of the minor isotopologue 35Cl37Cl+ . They agreed with Huberman on vibrational numbering of the ground state, and their analyses were also in agreement with a new analysis by Gharaibeh et al.[10] However, the vibrational numbering of the excited state seems to be modified. In 1990, Bramble and Hamilton[11] reported the first laser-induced fluorescence spectra of and analyzed many bands of the (υ ′ , 0) progression of the Ω = 3/2 component together with some extra bands arising from the perturbation.
In 2012, Gharaibeh et al.[10] studied the laser-induced fluorescence spectrum of jet-cooled with high sensitivity and rigorous vibrational and spin– orbit cooling. A full understanding of the A2Π 3/2– X2Π 3/2 band system was presented, including a complete treatment of the interaction between the A2Π u3/2 and B2Δ u3/2 states, based on the experimental spectrum and ab initio calculations. The (0, 0) band was identified for the first time. They studied two bands at high-resolution and obtained the accurate B values. For most of the low-resolution bands, they listed approximate upper state B values as well.
More information about the electronic states of has been derived from the photoelectron spectroscopy (PES).[7, 12, 13] In 2007, Li et al.[12] studied the isotopomer-resolved vibrational and spin– orbit energy structures of by one-photon zero kinetic energy (ZEKE) photoelectron spectroscopy. The precise values of the spin– orbit splitting for X2Π g were reported. Most recently, Mollet and Merkt[13] studied the X2Π g, A2Π u, B2Δ u, and states of by partially rotationally resolved pulsed-field-ionization zero-kinetic-energy (PFI-ZEKE) photoelectron spectroscopy. A large number of Franck– Condon-forbidden transitions to vibrational levels of the X2Π g (Ω = 3/2, 1/2) were observed. The potential energy function of was determined in a direct fit to the experimental data. Transitions to vibrational levels of the A2Π u3/2, B2Δ u3/2 states were identified by using the results of Ref. [10] and transitions to the upper spin– orbit component A2Π u1/2 (υ = 2– 6) were detected. Additionally, the A2Π u1/2 state was found to be perturbed by the 2Σ u1/2 state. Unfortunately, their data were too scarce to model this interaction.
In the present work, we study the rotational structure of the Ω = 3/2 component of the X2Π g, A2Π u, and B2Δ u3/2 states. Aiming to obtain the equilibrium rotational constants Be and α e of these states based on the experimental data, we reassign the vibrational quantum numbers for the previously observed bands and subsequently fit the rotational constants. Using the isotopic effect, the rotational spectroscopic constants for the minor isotopologues 35Cl37Cl+ and are predicted.
2. Theoretical details
Two methods are used to calculate the values of origin T0 for vibrational bands of the A2Π 3/2– X2Π 3/2 system of . The first method is to use the effective Hamiltonian matrix. The form of the matrix is shown in Fig. 8 of Ref. [10]. The diagonal energy terms are Eυ = Te + Gυ , where
The off-diagonal terms are , where the first term is the electronic spin– orbit interaction matrix element and the second term is a vibrational overlap.[14] The vibrational overlaps are calculated by using LeRoy’ s RKR1 2.0 and LEVEL 8.0 programs.[15, 16] Additionally, for calculating the matrix elements, we adopt the reported vibrational constants and also the spin– orbit coupling term .[10] The matrix is diagonalized and the eigenvalues are stored for calculating the values of origin T0 (υ ′ , υ ″ ) = Eυ ′ − Eυ ″ of vibrational bands. Note that the adopted vibrational constants and the interaction constant are fitted by using the band heads rather than band origins in Ref. [10], because the band origins cannot be determined accurately without full rotational analysis. However, it was argued that this introduces a little error (only 0.3 cm− 1– 0.5 cm− 1), which is well within the overall standard error for the least square analysis. As to the vibrational assignment, it is accurate enough. Moreover, the band heads replace the band origins in the second calculation method.
The second method is much simpler: there is no need to calculate the vibrational overlaps nor to diagonalize the matrices. Using the experimentally observed positions of the band heads, such as T0 (υ ′ , 0) of the (υ ′ , 0) bands, the values of band origin T0 (υ ′ , υ ″ ) can be evaluated by using the following formulas:
and
The vibrational constants (645.6 cm− 1) and (2.98 cm− 1) of [13] are adopted. In Ref. [10], 70 band heads of the species are listed, which all originate from the υ ″ = 0 level of the X2Π 3/2 state. The relevant upper levels are A2Π 3/2(υ ′ = 0– 39) and B2Δ u3/2(υ ′ = 0, 1, 3– 8, 10– 12, 14– 19, 21– 26, 28– 29, 31, 32). The extent of the upper levels and the accuracies of the band heads are sufficient for the purpose of this work.
Molecular chlorine has three isotopologues, 35Cl2, 35Cl37Cl, and 37Cl2 with relative abundances of 57.41%, 36.72%, and 5.87%, respectively.
The spectroscopic constants for two different isotopologues i and j have the following relationships:[17]
and
with
where μ i and μ j are the reduced mass values for isotopologues i and j. The values of ρ for the pairs 35Cl2 and 35Cl37Cl, and 35Cl2 and 37Cl2 are 0.98640 and 0.97261 respectively. The investigations of the isotopic effect in electronic band spectra lead to the determination of the correlated constants[18] and are helpful for discovering the new isotopologues of very small abundance.[19]
3. Results and discussion
The vibrational band origins of the A2Π 3/2– X2Π 3/2 and B2Δ u3/2– X2Π 3/2 are calculated by using the above two methods respectively and subsequently the observed bands reported in Refs. [6], [7], and [9] are reassigned. Listed in Table 1 are the origins with the vibrational assignments in a Deslandres table for the A2Π 3/2u– X2Π 3/2g system of . The vibrational assignments for the emission spectra of and 35Cl37Cl+ observed by Choi and Hardwick[9] are listed in Table 2. We agree on their ground state vibrational assignments but modify the vibrational numbering of the excited state A2Π 3/2. Moreover transitions to the B2Δ u3/2 state have also been assigned, and they are also listed in Table 2.
Table 1.
Table 1.
Table 1. Deslandres table for . All data are in units of cm− 1.
Table 2. Vibrational assignments for the emission spectra of and 35Cl37Cl+ observed by Choi and Hardwick[9].
Based on the unambiguous vibrational assignments of the states, some vibrational quantum numbers υ with the rotational constants Bυ are modified consequently, such as in Ref. [9]. The rotational constants of the X2Π 3/2g, A2Π 3/2u, and B2Δ 3/2u states for and 35Cl37Cl+ in the previous work are listed in Table 3. The constants Be and α e are fitted according to Bυ = Be − α e(υ + 1/2). The derived values of Be and α e of the X2Π 3/2g and A2Π 3/2u states for and 35Cl37Cl+ and those of the B2Δ 3/2u state for are listed in Table 4. The values of equilibrium internuclear distance Re for those three states of all the three isotopologues are determined from the values of Be and listed in Table 4. The derived rotational constants of the X2Π 3/2g and A2Π 3/2u state are in good agreement with those from the ab initio PECS in Ref. [10]. For , the derived value of Be is 0.1746 cm− 1 in comparison with the theoretical value of 0.1617407 cm− 1.[10]
Using Eq. (7), the rotational constants of the minor isotopologues 35Cl37Cl+ and are predicted and listed in Table 4 as well.
It should be mentioned that according to the calculated vibrational band origins, the bands at 16913, 17030, 17282, and 17323 cm− 1 would be assigned as (1, 6) (3, 7) and (2, 6) of and (2, 6) of 35Cl37Cl+ of the A2Π 3/2– X2Π 3/2 sub-system instead, which are tentatively assigned as (2, 7), (3, 7), and (4, 8) of and (3, 7) of 35Cl37Cl+ of the A2Π 1/2– X2Π 1/2.[20, 21] When modifying the vibrational quantum numbers of the bands, the experimental rotational constants in Refs. [20] and [21] are in good agreement with those in Ref. [10].
Table 3.
Table 3.
Table 3. Experimentally determined rotational constants of the X2Π 3/2g, A2Π 3/2u, and B2Δ 3/2u states. All data are in units of cm− 1.
Table 3. Experimentally determined rotational constants of the X2Π 3/2g, A2Π 3/2u, and B2Δ 3/2u states. All data are in units of cm− 1.
Table 4.
Table 4.
Table 4. Fitted rotational constants, the predicted rotational constants (in cm− 1), and the equilibrium internuclear distances of the X2Π 3/2g, A2Π 3/2u, and B2Δ 3/2u states.
35Cl37Cl+
This work
Previous work
This work
This work
Previous work
This work
Previous work
X2Π 3/2g
Be
0.269791(85)a
0.26950b
0.262648(14)
0.262503e
0.255216e
0.2707(11)c
α e
0.001665(27)
0.00164b
0.0016730(52)
0.001598e
0.001532e
0.0012(4)c
σ
0.00006
0.0000073
Re
1.891 Å
1.890 Å
1.891 Å
1.891 Å
A2Π 3/2u
Be
0.1903(14)
0.1897495d
0.1878(24)
0.1852e
0.1846241d
0.1800e
0.1794985d
α e
0.001770(63)
0.001375892d
0.00208(28)
0.0016988e
0.001320652d
0.001629e
0.001265973d
σ
0.0038
0.003
Re
2.251 Å
2.25 Å d
2.235 Å
2.251 Å
2.254 Å f
2.251 Å
2.254 Å f
B2Δ 3/2u
Be
0.1746(12)
0.1617407d
0.1699e
0.1573721d
0.1652e
0.1530031d
α e
0.001561(69)
0.001193303d
0.001498e
0.001146015d
0.001436e
0.001098608d
σ
0.003
Re
2.350 Å
2.44 Å d
2.350 Å
2.442 Å f
2.350 Å
2.442 Å f
Numbers in parentheses are in the units of the last quoted digits.
Calculated based on the constants of the previous work.
Table 4. Fitted rotational constants, the predicted rotational constants (in cm− 1), and the equilibrium internuclear distances of the X2Π 3/2g, A2Π 3/2u, and B2Δ 3/2u states.
However there is less experimental information about the A state Ω = 1/2 levels than about the Ω = 3/2 levels. Theoretically, the A2Π 1/2u levels above ∼ υ = 4 will be perturbed by levels of the state.[10] In the experiment, the jet-cooling only allows the Ω = 3/2– Ω = 3/2 transitions to be observed.[10] If more bands of the other half transitions Ω = 1/2– Ω = 1/2 could be observed, combined with the data reported in previous work, the data could be fitted to understand the whole structure of and the spin– orbit coupling constant A for the A2Π u state would precisely be obtained.
4. Conclusions
The vibrational and rotational structures of the A2Π 3/2, B2Δ u3/2, and X2Π 3/2 states of the three isotopologues of (, 35Cl37Cl+ , ) are studied. Using the previous experimentally observed positions of the band origins and the vibrational constants, [10, 13] we calculate the vibrational band origins of the A2Π 3/2– X2Π 3/2 and B2Δ u3/2– X2Π 3/2 systems and reassign the observed bands reported in Refs. [6], [7], and [9]. Some vibrational quantum numbers of the states are modified, and thus the experimental rotational constant Bυ are modified accordingly.
Based on the unambiguous vibrational assignments of the states, the values of Bυ reported in previous investigations are used to derive the constants Be and α e of the X2Π 3/2g and A2Π 3/2u states for and 35Cl37Cl+ and those of the B2Δ 3/2u state for . Then the rotational constants of the minor isotopologues 35Cl37Cl+ and are predicted based on the isotopic relations. Additionally, the values of equilibrium internuclear distance Re for those three states of all the three isotopologues are calculated.
Lefebvre-BrionH and FieldR W2004The Spectra and Dynamics of Diatomic MoleculesAmsterdam: Elsevier[Cited within:1]
15
Le RoyR J2004RKR1 2. 0: A computer program implementing the first-order RKR method for determining diatomic molecule potential energy functionsUniversity of Waterloo Chemical Physics Research Report No. CP-657R[Cited within:1]
16
Le RoyR J2007LEVEL 8. 0: A computer program for solving the radial Schrödinger equation for bound and quasibound levelsUniversity of Waterloo Chemical Physics Research Report No. CP-663[Cited within:1]
17
HerzbergG1950Molecular Spectra and Molecular Structure I: Spectra of Diatomic MoleculesNew Yorkvan Nostrand [Cited within:1]
... 5] It is difficult to assign the absolute vibrational quantum numbers to the observed bands because of the irregular vibrational internals and non-systematic chlorine isotopic effects resulting from the severely perturbed excited electronic state A2#cod#x03A0 ...
3
1966
0.0
0.0
... In 1966, Huberman[6] studied the emission spectrum and made the most comprehensive rotational assignment of the observed ...
... [6], [7], and [9] are reassigned ...
... [6], [7], and [9] ...
4
1984
0.0
0.0
... In 1989, Tuckett and Peyerimhoff[7] observed the emission spectrum in the supersonic expansion, which allowed them to make a fuller analysis due to the cold Doppler-free spectrum ...
... [7,12,13] In 2007, Li et al ...
... [6], [7], and [9] are reassigned ...
... [6], [7], and [9] ...
1
1989
0.0
0.0
... X emission spectrum by Choi and Hardwick,[8] and four bands were unambiguously assigned as A2#cod#x03A0 ...
5
1991
0.0
0.0
... Later in 1991, they[9] reported more bands of the #cod#x03A9 ...
... [6], [7], and [9] are reassigned ...
... observed by Choi and Hardwick[9] are listed in Table 2 ...
... [9] ...
... [6], [7], and [9] ...
13
2012
0.0
0.0
... [10] However, the vibrational numbering of the excited state seems to be modified ...
... [10] studied the laser-induced fluorescence spectrum of jet-cooled with high sensitivity and rigorous vibrational and spin#cod#x2013 ...
... [10] and transitions to the upper spin#cod#x2013 ...
... [10] ...
... [10] The matrix is diagonalized and the eigenvalues are stored for calculating the values of origin T0 (#cod#x03C5 ...
... [10], because the band origins cannot be determined accurately without full rotational analysis ...
... [10], 70 band heads of the species are listed, which all originate from the #cod#x03C5 ...
... [10] ...
... [10] ...
... [10] ...
... [10] In the experiment, the jet-cooling only allows the #cod#x03A9 ...
... [10] If more bands of the other half transitions #cod#x03A9 ...
... Using the previous experimentally observed positions of the band origins and the vibrational constants,[10,13] we calculate the vibrational band origins of the A2#cod#x03A0 ...
1
1990
0.0
0.0
... In 1990, Bramble and Hamilton[11] reported the first laser-induced fluorescence spectra of and analyzed many bands of the (#cod#x03C5 ...
2
2007
0.0
0.0
... [7,12,13] In 2007, Li et al ...
... [12] studied the isotopomer-resolved vibrational and spin#cod#x2013 ...
4
2013
0.0
0.0
... [7,12,13] In 2007, Li et al ...
... Most recently, Mollet and Merkt[13] studied the X2#cod#x03A0 ...
... 1) of [13] are adopted ...
... Using the previous experimentally observed positions of the band origins and the vibrational constants,[10,13] we calculate the vibrational band origins of the A2#cod#x03A0 ...
1
2004
0.0
0.0
... [14] The vibrational overlaps are calculated by using LeRoy#cod#x2019 ...
1
2004
0.0
0.0
... [15,16] Additionally, for calculating the matrix elements, we adopt the reported vibrational constants and also the spin#cod#x2013 ...
1
2007
0.0
0.0
... [15,16] Additionally, for calculating the matrix elements, we adopt the reported vibrational constants and also the spin#cod#x2013 ...
1
1950
0.0
0.0
... The spectroscopic constants for two different isotopologues i and j have the following relationships:[17] ...
1
2014
0.0
0.0
... The investigations of the isotopic effect in electronic band spectra lead to the determination of the correlated constants[18] and are helpful for discovering the new isotopologues of very small abundance ...
1
2009
0.0
0.0
... [19] ...
2
2005
0.0
0.0
... [20,21] When modifying the vibrational quantum numbers of the bands, the experimental rotational constants in Refs ...
... [20] and [21] are in good agreement with those in Ref ...
2
2008
0.0
0.0
... [20,21] When modifying the vibrational quantum numbers of the bands, the experimental rotational constants in Refs ...
... [20] and [21] are in good agreement with those in Ref ...
Study on the A2Π3/2u, B2Δ3/2u, and X2Π3/2g states of including its isotopologues
[Wu Linga), You Su-Pinga), Shao Xu-Pingb), Chen Gang-Jina), Ding Ninga), Wang You-Meia), Yang Xiao-Hua†b)]

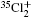
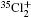

 ]
]