Xiao Hong-Jun, Shen Cheng-Min, Shi Xue-Zhao, Yang Su-Dong, Tian Yuan, Lin Shao-Xiong, Gao Hong-Jun. Synthesis of graphene-supported monodisperse AuPd bimetallic nanoparticles for electrochemical oxidation of methanol. Chinese Physics B, 24(7): 078109
Permissions
Synthesis of graphene-supported monodisperse AuPd bimetallic nanoparticles for electrochemical oxidation of methanol
Xiao Hong-Juna),b), Shen Cheng-Mina),b), Shi Xue-Zhaoa), Yang Su-Donga), Tian Yuana),b), Lin Shao-Xionga), Gao Hong-Juna),b)†
Beijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
University of Chinese Academy of Sciences, Beijing 100049, China
*Project supported by the National Natural Science Foundation of China (Grant No. 61335006) and the National Basic Research Program of China (Grant No. 2013CBA01603).
Abstract
Monodisperse AuPd bimetallic nanoparticles (NPs) with different compositions are synthesized by using oleylamine (OAm) as reducing reagent, stabilizer, and solvent. To obtain AuPd solid solution NPs, Pd–OAm and Au–OAm precursors are firstly prepared by mixing OAm with Palladium (II) acetylacetonate (Pd(acac)2) and HAuCl4, respectively. Then Pd–OAm and Au–OAm precursor solutions are injected into a hot oleylamine solution to form AuPd NPs. The size of these NPs ranges from 6.0 to 8.0 nm and the composition is controlled by varying the precursor ratio. The AuPd NPs are loaded onto reduced graphene oxide (RGO) sheets to make catalysts. Alloy NPs show high electrocatalytic activity and stability toward methanol oxidation in the alkaline media. Their catalytic activity for methanol oxidation is found to be dependent on the NP composition. As the Pd component increases, the peak current densities during the forward scan gradually increase and reach the maximum at AuPd2. The enhancement of alloy NPs for methanol oxidation can be attributed to a synergistic effect of Au and Pd on the surface of alloy NPs.
Nanomaterials are playing an important role in current fuel cell technologies.[1] Pt nanoparticles (NPs) have been extensively studied and widely used as catalysts for various fuel cells, such as hydrogen and direct methanol fuel cells.[2] However, the use of Pt in fuel cells has been limited by its high cost and low tolerance for CO poison.[2, 3] Therefore, it is important to develop alternative non-Pt catalysts with comparable catalytic properties.[4, 5] It has been found that Pd is a promising candidate due to its unique catalytic properties in many organic reactions and industrial productions.[6] As a plus, Pd costs less than Pt and is 50 times more abundant on earth.[7] Recently, great effort has been devoted to make Pd nanomaterials through different routes for fuel cell related reactions.[8– 11] Pd nanocatalysts have shown high electroactivity and stability for direct alcohol and formic acid oxidations.[12– 15] These studies on pure Pd catalysts inspired a great interest to make Pd-based bimetallic NPs and investigate their catalytic activities.[16] Previous studies on Pt-based bimetallic catalysts have shown that varying the NPs’ composition can significantly change their catalytic properties.[17– 21] With different compositions, the surface of bimetallic NPs shows different electronic properties, and thus a variation of catalysis efficiency can be observed.[22– 24] These findings have encouraged the similar studies on Pd-based bimetallic nanoparticle systems. To date, AuPd bimetallic nanoparticles in alloy and core/shell structures have been successfully prepared using a wet-chemical method.[25– 32] However, to control the composition of colloid AuPd within a retainable size range and further to evaluate the dependence of their catalytic activities on composition have been reported rarely.
In order to maximize the electrocatalytic activity of AuPd alloy NPs, catalyst NPs need to be dispersed on the surface of suitable materials. Among the catalyst support, graphene has attracted considerable attention as a high performance catalyst support material for fuel cell reactions.[33– 35] This derives from the high conductivity, large specific surface area, chemical and mechanical stability of graphene.[36]
In this paper, we report a simple wet-chemical synthesis of AuPd bimetallic nanoparticles with controllable compositions and their electrochemical catalytic activities toward methanol oxidation. The AuPd nanoparticles were synthesized using oleylamine (OAm) as reducing reagent, surfactant, and solvent.[31, 37, 38] Pd– OAm and Au– OAm precursors were first prepared by dissolving Pd (acac)2 and HAuCl4 in oleylamine. Then AuPd NPs were produced by injecting the precurosrs into oleylamine solution at 120 ° C to produce AuPd NPs. The composition of nanoparticles was controlled by varying the feeding ratio of Au and Pd precursors. The inductively couple plasma atomic emission spectroscopy (ICP– AES) and energy-dispersive x-ray (EDX) spectra analyses show that the composition of as-prepared AuPd NPs was determined by the ratio of precursors. The high-resolution TEM (HRTEM) and XRD analyses reveal that these AuPd NPs show polycrystalline crystal properties and are indeed well-mixed solid solution. The surface plasma absorption of Au is diminished as the Pd component is increased. Further electrochemical studies indicate that these AuPd NPs loaded onto RGO exhibit an enhancement in catalytic activity for methanol oxidation
2. Experimental section
2.1. Materials
Pd(acac)2 (99%), oleylamine (80%– 90%) and hexane were purchased from Sigma-Aldrich. Ethanol (99%), and HAuCl4· 4H2O (analyst degree) were obtained from Beijing Chemical Reagent Com. All reagents were used without further purification.
2.2. Synthesis of AuPd NPs
In a 10-mL vial, 0.25 mmol HAuCl4· 4H2O was dissolved in 2.0 mL oleylamine and 2.0 mL hexane to form a red solution. In another 10-mL vial, 0.25 mmol Pd(acac)2 was dissolved in 3 mL oleylamine at 50 ° C to obtain a light yellow solution. Then 15 mL oleylamine was added into a 100-mL flask and heated to 120 ° C for 30 min under nitrogen atmosphere. The Pd– OAm solution was first injected into 120 ° C oleylamine solution with vigorous stirring. After 5 min, the color of solution gradually changed to gray black, and then 4 mL HAuCl4· 4H2O/hexane/OAm solution was rapidly injected and kept at 120 ° C for 120 min. The solution was cooled down to room temperature, 30 mL of ethanol was added, and the product was separated by centrifuge (6000 rpm for 3 min). The black precipitant was washed by acetone three times. The product was then dispersed in hexane. AuPd NPs with different composition were synthesized by changing molar ratio of precursors: molar ratios of HAuCl4· 4H2O to Pd(acac)2 were 2:1 and 1:2, respectively, which correspond to Au2Pd and AuPd2 NPs.
2.3. Preparation of reduced graphene oxide (RGO) sheets
The graphite oxide (GO) was synthesized from natural graphite powder according to a method reported in Ref. [39]. 20 mg GO powder was loaded into a quartz boat, then quartz boat was put into a horizontal tube furnace. When the system was pumped below 10 Pa, 100 sccm H2/Ar mixed gas was introduced into the system, and the furnace was heated to 1000 ° C at a rate of 10 ° C/min. After reaction for 2 h, the furnace was cooled down to room temperature. RGO sheets were prepared (Fig. A1).
2.4. Preparation of RGO supported NPs
A typical preparation of Au– Pd/RGO hybrids is as follows: in a 20-mL hexane solution containing 10 mg alloy NPs, 10 mg RGO was added. Then this colloidal solution was sonicated for 4 h. The cubic Au– Pd/RGO hybrids were finally separated by centrifugation, and the carbonsupported NPs were treated with 20 mL acetic acid at 70 ° C for 10 h. The suspension was cooled down to room temperature, 30 mL ethanol was added, and the mixture was centrifuged at 8000 rpm for 8 min. This procedure was repeated twice. The catalysts were finally dispersed in deionized water to obtain a 2.0 mg/mL solution. For comparison, the XC-72 carbon black supported alloy NPs were prepared using the same process.
2.5. Characterizations
The XRD patterns were recorded using Rigaku x-ray diffractometer with Cu Kα radiation (λ = 1.5406 nm). The morphology of NPs was investigated by an HITACH I8100 transmission electron microscopy (TEM) with an operating voltage of 160 kV. High-resolution TEM (HRTEM) images were obtained on a JEOL 2010 at 200 kV. The UV– Vis spectrum was recorded on a Cary 1E ultraviolet-visible spectrometer using 1-cm quartz cuvettes. The composition of NPs was determined by IRIS Intrepid II ICP– AES.
2.6. Electrochemical measurements
The cyclic voltammograms were recorded on a CHI 660D electrochemical work station (Shanghai, China). A standard three-electrode system was used. A glassy carbon (GC) electrode with a diameter of 5.0 mm was used as the working electrode. A platinum wire and a saturated calomel electrode (SCE) were used as counter electrode and reference electrode, respectively. The electrochemical oxidation of methanol measurement was performed in 0.1 M KOH + 1.0 M CH3OH solution. The electrolyte solutions were degassed by nitrogen for 30 min before measurement. All of the electrochemical evaluations were performed at room temperature.
3. Results and discussion
Monodisperse AuPd NPs with different compositions were synthesized by using oleylamine as reducing reagent and solvent at 120 ° C. The morphology and structure of different composition AuPd NPs were characterized by TEM. Figure 1(a) shows a typical TEM image of AuPd NPs with a 1:1 Au/Pd ratio. The average diameter of these NPs is about 6.9 nm. The HRTEM image shows the typical multiple-twined structure, which was usually found in Au-based bimetallic NPs.[38] The corresponding SAED pattern (the inset of Fig. 1(b)) of AuPd NPs shows a single phase of fcc diffraction features, excluding the phase separation between Au and Pd.[40]
Fig. 1. A typical TEM image (a) and an HRTEM image (b) of as-synthesized AuPd nanoparticles (Au/Pd ratio: 1). The inset of HRTEM image shows the corresponding SAED pattern, indicating a fcc structure of AuPd.
The composition of AuPd NPs was varied from Au2Pd to AuPd2 (Fig. A2). So-produced AuPd nanoparticles were examined by ICP and EDX, and it was found that the nanoparticle composition is directly proportional to the feeding ratio of precursors (Fig. A3).
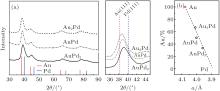
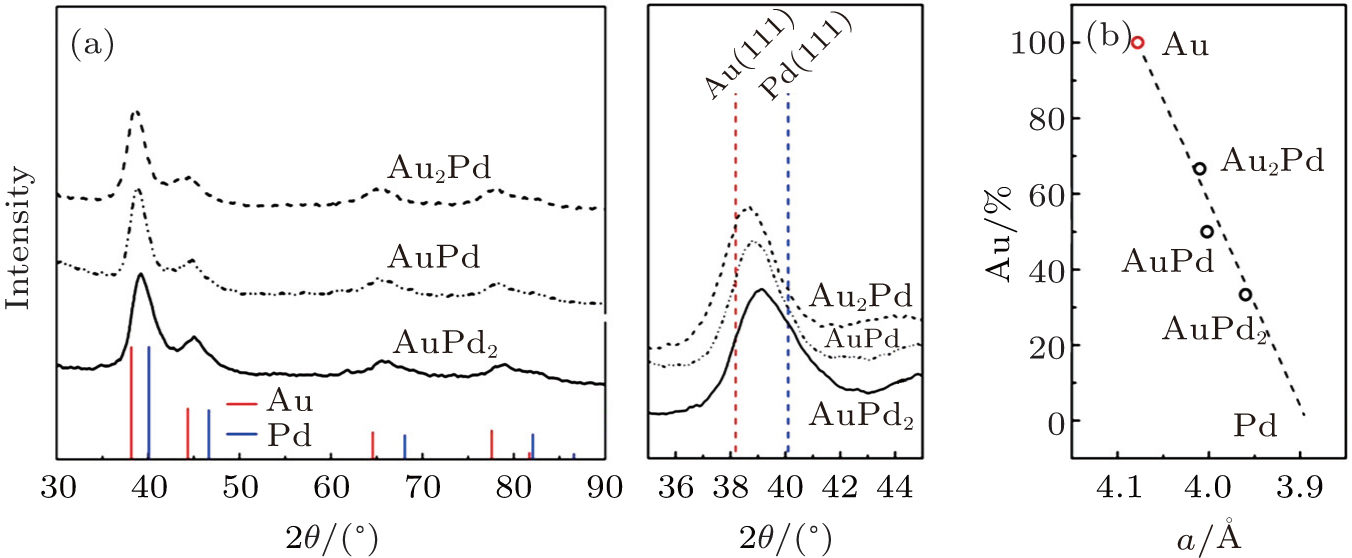
To further analyze the crystal structure of these AuPd bimetallic NPs, XRD patterns were recorded (Fig. 2(a)). All the XRD patterns of AuPd nanoparticles show typical fcc structure with four major feature peaks of (111), (200), (220), and (311). These diffraction peaks are located between the standard peaks of Au and Pd and shift from the Au side to Pd side regularly with the decrease of Au component. The calculated lattice parameters of these AuPd NPs are 4.010, 4.002, and 3.960 Å for Au2Pd1, Au1Pd1, and Au1Pd2, respectively. The fitted curve between the lattice parameters of Au (4.078 Å ) and Pd (3.891 Å ) was linear, indicating that the two metals are well-mixed.
Fig. 2. XRD patterns (a) and calculated lattice parameters (b) of as-synthesized AuPd NPs. All the AuPd NPs show a single phase of fcc structure and their diffraction peaks shift from Au standard (PDF#65-2870) to Pd standard (PDF#65-2867) as Au composition (Au%) decreases. The calculated lattice parameters of Au/Pd ratio are linear.
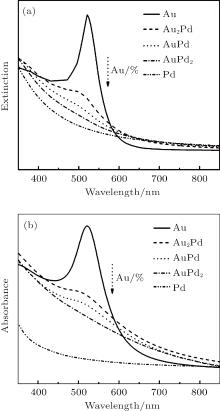
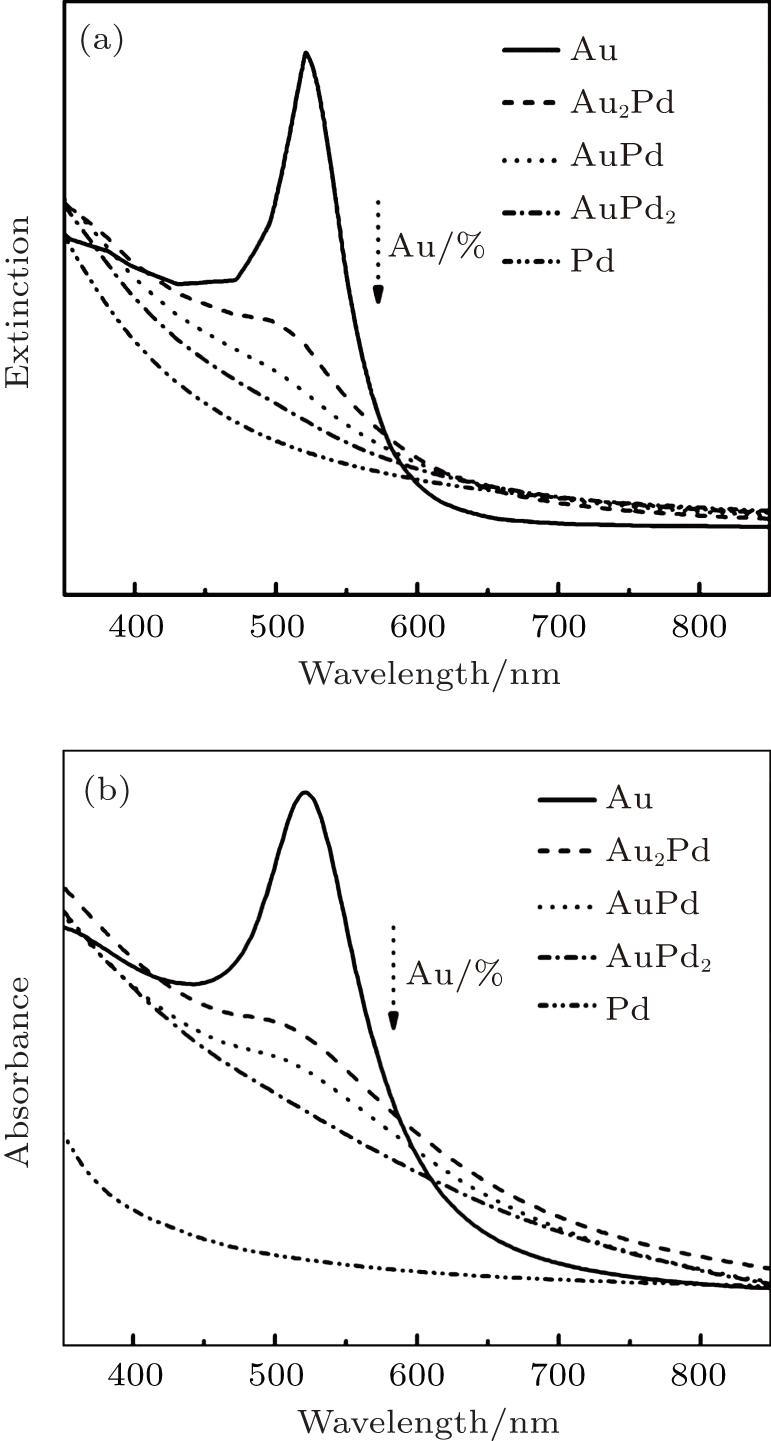
Figure 3(a) shows the calculated UV– Vis spectra of well-mixed AuPd NPs with different compositions. The model of AuPd is built on monodisperse spheres with a diameter of 7 nm. The calculation was based on Mie theory in the dipole approximation, in which a linear combination of the dielectric constant of Au and Pd according to the respective Au molar fraction XAu is assumed.[41] The dielectric function (ε AuPd) of AuPd particle is in the form of
where x is the molar fraction of Au, and ε is the dielectric function constant of each metal.[42] The calculated spectra show a dependence of absorption intensity on Au fractions. With the decrease in Au component (Au%), the absorption of AuPd spheres decreases regularly and completely diminishes as Au% is no more than 33%. Figure 3(b) shows the experimental spectra recorded from as-synthesized AuPd NPs. It is clear that the absorption peak of NPs drops regularly as the Au% decreases from 100% to 50% and completely disappears as Au% is less than 33%. Compared with the calculated spectra, the experimental spectra show slight variations on peak position and width, which is due to the variation of nanoparticle size dispersity. The experiment spectra are consistent with the calculated ones, and further indicate that the synthesis gives well-mixed AuPd bimetallic NPs.
Fig. 3. Calculated UV– Vis spectra (a) and experimental spectra (b) of AuPd NPs as a function of Au composition (Au%). The calculated spectra are recorded by modelling monodisperse 7-nm spherical particles through Mie theory. The complex dielectric function of AuPd is calculated as a function of the molar fraction of Au and Pd components.
The morphologies of the RGO nanosheet-supported different composition AuPd alloy NPs were characterized using TEM. Figure 4 shows the TEM images of different component Au– Pd/RGO composites. It can be seen that the RGO does not degrade the quality of the AuPd alloy NPs after alloy NPs are deposited on the surface of RGO. The AuPd alloy NPs have uniform size and are well-dispersed on the support material surface. Based on the TEM results, we see that the size and shape of AuPd alloy NPs do not change during the RGO loading process. For comparison, AuPd alloy was loaded on the XC-72 carbon black using the same method (Fig. A4).
Fig. 4. TEM images of Au– Pd/RGO catalysts: (a) Au2Pd; (b) AuPd; and (c) AuPd2. The results indicate that AuPd NPs are dispersed uniformly on the surface of carbon supports.
The loading mass of the AuPd NPs was kept at 50%. The acetic acid treatment was employed for surfactant removal.[8] 1.0 mL as-prepared Au– Pd/RGO catalyst NPs were mixed with 1.0 mL 0.05% Nafion under sonication for 20 min. 20 μ L Au– Pd/RGO– Nafion mixed solution was dropped on a glassy carbon electrode and dried slowly at room temperature to form a catalyst thin film on electrode. The electrochemical measurement was carried out at room temperature using a standard threeelectrode system. The cyclic voltammetric characteristics of Au– Pd/RGO NPs were tested in 0.1 M KOH solution at a scan rate of 50 mV/s (Fig. 5). It is seen clearly that the oxygen reduction peaks of AuPd alloy NPs with different compositions are located between those of Pd NPs and Au NPs. The oxygen reduction peaks of AuPd alloy NPs gradually shift toward negative potential with increasing Pd content in alloy NPs, revealing different component alloy forms of the Au– Pd atoms on the NP surface.[43– 48]
Fig. 5. Cyclic voltammogram of RGO sheets supported NPs in 0.1 M KOH solution at a scan rate of 50 mV/s.
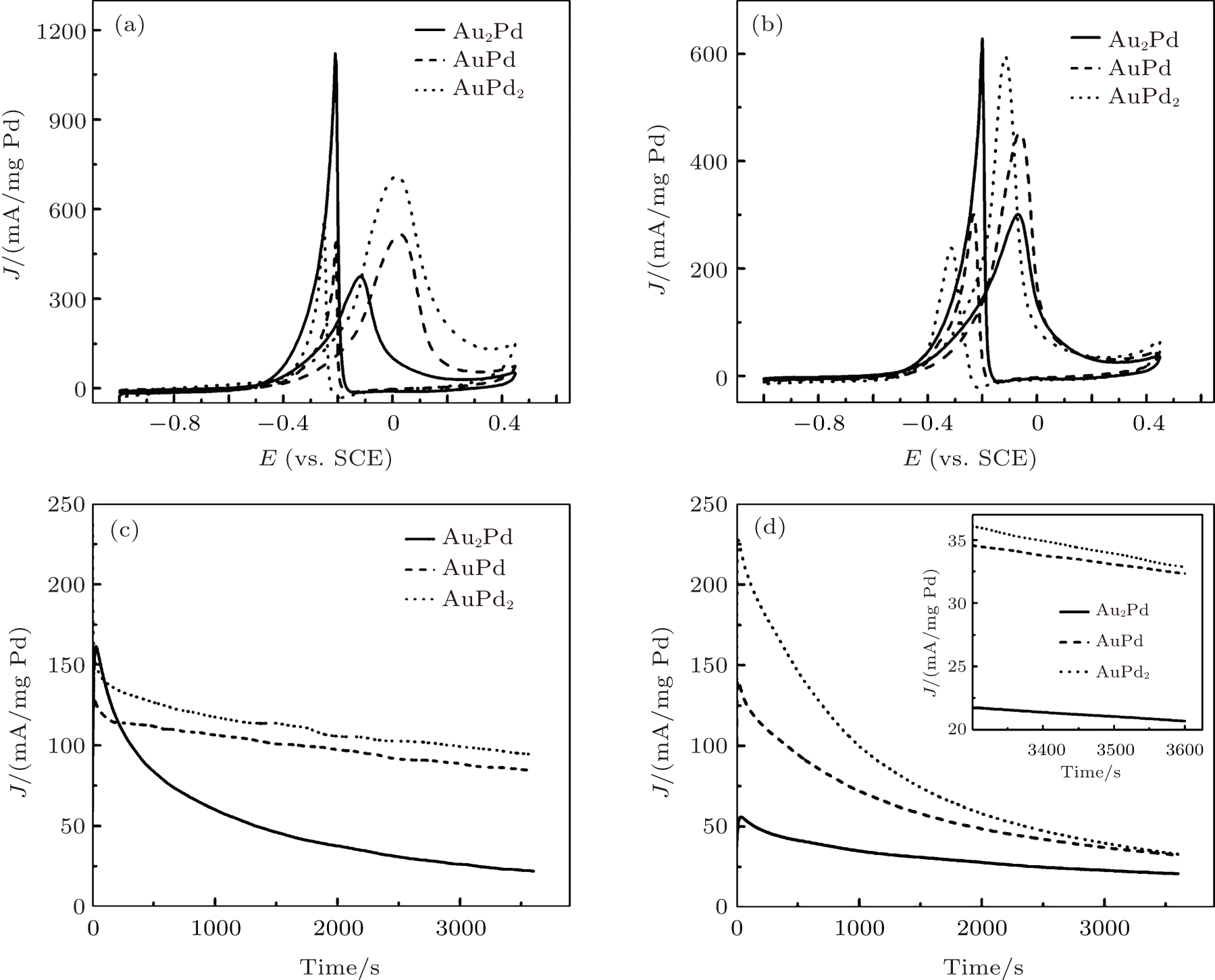
Pd-based nanostructures are known to have a good electrocatalytic activity for methanol oxidation in alkaline media.[5] Figure 6(a) shows the cyclic voltammogram curves of Au– Pd/RGO NPs in 0.1 M KOH and 1.0 M methanol. Two oxidation peaks were found on the forward and backward scans, corresponding to oxidation of methanol and removal of carbonaceous species not completely oxidized in the forward scan, respectively.[44] The current densities were normalized by corresponding the weight of the Pd component in AuPd NP catalysts in the electrode. Their current densities at the forward scan peaks are 379.6, 518.1, and 711.6 mA/mg Pd for Au2Pd, AuPd, and AuPd2, respectively. As the Pd component increases, the current densities at the forward scan peaks increase and reach the maximum at AuPd2. These results indicate that the catalytic activity enhancement of AuPd alloy NPs is obviously composition-dependent. The highest specific activity was found at an optimal Au/Pd ratio of 1/2. The similar enhancement was also found on AuPd nanodendrites for methanol oxidation in alkaline.[44] It is known that Au is not active for methanol oxidation in acid, but active in alkaline media.[49– 51] However, its specific activity is low and not comparable with Pd. The enhancement on these AuPd alloy NPs for methanol oxidation probably can be attributed to a synergistic effect of Au and Pd on the surface of alloy NPs.[52] The ability of Au to enhance oxygen species probably enables the improvement on the kinetics of some intermediate products removal.[50, 51, 53] Comparing to results in Fig. 6(b), the current density of Au– Pd/RGO for methanol oxidation is nearly 1.27 (Au2Pd), 1.14 (AuPd), and 1.20 (AuPd2) times higher than that of Au– Pd/XC-72 catalysts (Fig. 4(b)), respectively. These results indicate that the catalytic activity of Au– Pd/RGO hybrids is considerably higher than that of Au– Pd/CX-72. The enhancement in catalytic activity of Au– Pd/RGO NPs for methanol oxidation is possibly attributed to large surface area and good electrical conductivity of RGO sheets. On the other hand, various oxygen functional groups (e.g., hydroxyl and epoxy) can be formed on the basal planes of graphene when GO was reduced into RGO.[54] Sharma et al. reported that the oxygen groups on the RGO are important to remove carbonaceous specific adsorbed on the active Pt sites via the bifunctional mechanism.[55]
Fig. 6. Cyclic voltammograms of different composition AuPd catalysts in 0.1 M KOH + 1.0 M CH3OH solution at a scan rate of 50 mV/s. (a) AuPd/RGO; (b) AuPd/XC-72. Chronoamperometric curves of different composition AuPd catalysts in 0.1 M KOH + 1.0 M CH3OH solution at − 0.20 V (vs. SCE); (c) AuPd/RGO; and (d) AuPd/XC-72.
The chronoamperometric curves of Au– Pd/RGO alloy NPs for methanol oxidation was recorded at − 0.20 V versus SCE (Fig. 6(c)). It can be seen that the Au– Pd/RGO alloy NPs show activity with Pd component increase in AuPd NPs. The electrooxidation current at 3600 s on Au– Pd/RGO electrode was 21.7, 84.1, and 94.5 mA/mg Pd for Au2Pd, AuPd, and AuPd2, respectively. From Figs. 6(c) and 6(d), we can find that the current densities, activity, and durability of different composition AuPd NPs loaded on the surface of RGO sheets are higher than that of AuPd NPs loaded on XC-72. The currents of Au– Pd/RGO NPs are 2– 3 times higher than that of Au– Pd/XC-72 catalysts. These results indicate that Au– Pd/RGO has a higher stability toward methanol. It further reveals the high electrocatalytic activity and stability of different composition of Au– Pd/RGO for methanol oxidation.
4. Conclusions
Monodisperse AuPd bimetallic NPs with different compositions have been successfully prepared using oleylamine (OAm) as reducing reagent and solvent. The size of as-prepared AuPd NPs ranges from 6.0 to 8.0 nm. Their composition is controlled by varying the molar ratio of precursors. The AuPd alloy NPs are characterized using HRTEM, EDX, XRD, UV– Vis and ICP– AES. These results prove that well-mixed AuPd alloy is formed in these NPs. So-produced AuPd bimetallic NPs exhibit enhanced catalytic activities for the electrochemical oxidation of methanol. The current densities, activity, and durability of different composition AuPd NPs loaded on the surface of RGO sheets are much higher than that of AuPd NPs loaded on XC-72. The experimental results reported here are encouraging for developing non-Pt catalysts as well as Pd-based bimetallic NPs for fuel cell reactions.
... [1] Pt nanoparticles (NPs) have been extensively studied and widely used as catalysts for various fuel cells, such as hydrogen and direct methanol fuel cells ...
2
2009
0.0
0.0
... [2] However, the use of Pt in fuel cells has been limited by its high cost and low tolerance for CO poison ...
... [2,3] Therefore, it is important to develop alternative non-Pt catalysts with comparable catalytic properties ...
1
2006
0.0
0.0
... [2,3] Therefore, it is important to develop alternative non-Pt catalysts with comparable catalytic properties ...
1
2005
0.0
0.0
... [4,5] It has been found that Pd is a promising candidate due to its unique catalytic properties in many organic reactions and industrial productions ...
2
2009
0.0
0.0
... [4,5] It has been found that Pd is a promising candidate due to its unique catalytic properties in many organic reactions and industrial productions ...
... [5] Figure#cod#x00A0 ...
1
2005
0.0
0.0
... [6] As a plus, Pd costs less than Pt and is 50 times more abundant on earth ...
1
2010
0.0
0.0
... [7] Recently, great effort has been devoted to make Pd nanomaterials through different routes for fuel cell related reactions ...
2
2009
0.0
0.0
... [8#cod#x2013 ...
... [8] 1 ...
1
2009
0.0
0.0
1
2008
0.0
0.0
1
2006
0.0
0.0
... 11] Pd nanocatalysts have shown high electroactivity and stability for direct alcohol and formic acid oxidations ...
1
1996
0.0
0.0
... [12#cod#x2013 ...
1
2006
0.0
0.0
1
2007
0.0
0.0
1
2008
0.0
0.0
... 15] These studies on pure Pd catalysts inspired a great interest to make Pd-based bimetallic NPs and investigate their catalytic activities ...
1
2009
0.0
0.0
... [16] Previous studies on Pt-based bimetallic catalysts have shown that varying the NPs#cod#x2019 ...
1
2009
0.0
0.0
... [17#cod#x2013 ...
1
1999
0.0
0.0
1
2005
0.0
0.0
1
2013
0.0
0.0
1
2010
0.0
0.0
... 21] With different compositions, the surface of bimetallic NPs shows different electronic properties, and thus a variation of catalysis efficiency can be observed ...
1
2007
0.0
0.0
... [22#cod#x2013 ...
1
2009
0.0
0.0
1
2010
0.0
0.0
... 24] These findings have encouraged the similar studies on Pd-based bimetallic nanoparticle systems ...
1
1998
0.0
0.0
... [25#cod#x2013 ...
1
2003
0.0
0.0
1
2004
0.0
0.0
1
2005
0.0
0.0
1
2007
0.0
0.0
1
2008
0.0
0.0
1
2008
0.0
0.0
... [31,37,38] Pd#cod#x2013 ...
1
2008
0.0
0.0
... 32] However, to control the composition of colloid AuPd within a retainable size range and further to evaluate the dependence of their catalytic activities on composition have been reported rarely ...
1
2014
0.0
0.0
... [33#cod#x2013 ...
1
2014
0.0
0.0
1
2014
0.0
0.0
... 35] This derives from the high conductivity, large specific surface area, chemical and mechanical stability of graphene ...
1
2012
0.0
0.0
... [36] ...
1
2009
0.0
0.0
... [31,37,38] Pd#cod#x2013 ...
2
2010
0.0
0.0
... [31,37,38] Pd#cod#x2013 ...
... [38] The corresponding SAED pattern (the inset of Fig ...
1
1958
0.0
0.0
... [39] ...
1
2010
0.0
0.0
... [40] ...
1
1996
0.0
0.0
... [41] The dielectric function (#cod#x03B5 ...
1
1972
0.0
0.0
... [42] The calculated spectra show a dependence of absorption intensity on Au fractions ...
1
2011
0.0
0.0
... [43#cod#x2013 ...
2
2010
0.0
0.0
... [44] The current densities were normalized by corresponding the weight of the Pd component in AuPd NP catalysts in the electrode ...
... [44] It is known that Au is not active for methanol oxidation in acid, but active in alkaline media ...
1
2006
0.0
0.0
1
2009
0.0
0.0
1
1972
0.0
0.0
1
2001
0.0
0.0
... 48] ...
1
2008
0.0
0.0
... [49#cod#x2013 ...
1
2003
0.0
0.0
... [50,51,53] Comparing to results in Fig ...
2
2007
0.0
0.0
... 51] However, its specific activity is low and not comparable with Pd ...
... [50,51,53] Comparing to results in Fig ...
1
2007
0.0
0.0
... [52] The ability of Au to enhance oxygen species probably enables the improvement on the kinetics of some intermediate products removal ...
1
2008
0.0
0.0
... [50,51,53] Comparing to results in Fig ...
1
2013
0.0
0.0
... [54] Sharma et al ...
1
2010
0.0
0.0
... [55] ...
Synthesis of graphene-supported monodisperse AuPd bimetallic nanoparticles for electrochemical oxidation of methanol
[Xiao Hong-Juna),b), Shen Cheng-Mina),b), Shi Xue-Zhaoa), Yang Su-Donga), Tian Yuana),b), Lin Shao-Xionga), Gao Hong-Juna),b)†]







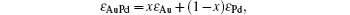


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 ]
]