
{kind=link}
{kind=link}
{kind=link}
Adsorption of glycine on diamond (001): Role of bond angle of carbon atoms*
[Li Lina) , Xu Jingb)†  , Xu Li-Fang
, Xu Li-Fanga) , Lian Chao-Shenga) , Li Jun-Jiea) , Wang Jian-Taoa) , Gu Chang-Zhia)‡, c) ]
, Xu Li-Fang]
|
|

 , Xu Li-Fang
, Xu Li-Fang
†Corresponding author. E-mail: xj2005@ruc.edu.cn
‡Corresponding author. E-mail: czgu@iphy.ac.cn
*Project supported by the National Natural Science Foundation of China (Grant Nos. 51272278, 91323304, 10774177, and 11374341), the National Basic Research Program of China (Grand No. 2009CB930502), the Knowledge Innovation Project of Chinese Academy of Sciences (Grand No. KJCX2-EW-W02), the Fundamental Research Funds for the Central Universities of Ministry of Education of China, and the Research Funds of Renmin University of China.
The adsorption behaviors of glycine on diamond (001) are systematically investigated by first-principles calculations. We have considered all possible adsorption configurations without a surface dangling bond and give a quantitative analysis for the relationship between the deviation of carbon bond angle and adsorption energy. We found that a smaller distortion of carbon covalent bond angle results in a more stable adsorption structure, and the most stable adsorption has a benzene-ring-like structure with the highest adsorption energy of 5.11 eV per molecule and the minimum distortion of carbon covalent bond angle.
With the development of nanotechnology, a new research field emerges, in which different materials with disparate mechanical and electronic properties are brought together to prepare new multifunctional devices.[1] It is believed that the combination of bio-molecules and current semiconductor technology could produce new ultrasensitive and hot-active synthetic components.[2] Nevertheless, the key to this is to make clear how bio-molecules adsorb on materials surface and how the surface properties transform after adsorption.
As a burgeoning semiconductor material, diamond presents unique physics and mechanical properties. With a wide indirect band gap of 5.5 eV, it has a high thermal conductivity and a broad optical transparency.[3, 4] Hence, the devices made using diamond will possess more outstanding characters under extreme conditions.[4] Because of the excellent bio-compatibility, diamond already begins to be used in bio-sensors.[5] Therefore, it is natural to consider combining biomolecule and diamond to fabricate multifunctional devices.
As the fundamental units of proteins, amino acids are interesting adsorbates for the studies of the interactions between biomolecules and solid surfaces. These organic molecules show a wide variety of charge, polarity, water interaction, and proton exchange. They are all made up with one amino group and one carboxyl group as common features, but with a unique functional group in the side chains. They are the basic units of complex biomolecule containing polypeptides and proteins. So amino acids can be used as model systems for studying biomolecule– surface interaction.
The adsorptions of glycine on solid surfaces, such as copper, [6– 9] gold, [10] silicon, [11] silica, [12] germanium, [13] etc. have been widely studied. Odbadrakh et al. preliminarily discussed the interactions of glycine and diamond (001).[14] However, some of their adsorption structures are not stable adsorption configurations since dangling bonds still exist. As a consequence, the surface band gaps vanished in some of their adsorption structures.
In this paper, we have intensively studied the adsorption behaviors of glycine on diamond (001) with first-principles calculations and acquired stable adsorptions with eliminating surface dangling bonds. All possible interaction sites of glycine were considered and the most stable adsorption structure was found in a benzene-ring-like geometry with the maximum adsorption energy of 5.11 eV per molecule. Particularly, the bond angles of carbon atoms play a key role for adsorption in this system. Further analysis is also given, which evidences that the smallest deviation of bond angle has the largest adsorption energy and vise verse.
The adsorptions of glycine on diamond (2× 1)-(001) surface were studied by first-principles calculations based on density functional theory. The Vienna ab initio simulation packages (VASP)[15], [16] with PBE functionals[17] and non-normconserving ultrasoft pseudopotentials[18] with a planewave cutoff of 350 eV was used. The systems were chosen as periodic supercells consisting of eight atomic layers for the diamond (001) surface with an equivalent number of vacuum layers. Throughout the calculation, the bottom layer of the structure was saturated with hydrogen atoms and kept frozen. Calculations were performed using the optimized surface structures, which consisted of the equivalent of 2× 4 surface dimers. The glycine molecule was optimized separately and then placed above the diamond surface. The entire system was then optimized using the conjugate gradient (CG) algorithm. The molecule and surface structures were considered to be equilibrated when the Hellmann– Feynman forces were less than 0.01 eV/Å . Brillouin zone integration for the surface band structure is performed using sets corresponding to 20 k points in the full (1× 1) surface Brillouin zone.
The adsorption energy of glycine on the (001) surface is given by

where Etotal is the total energy of the optimized adsorption structure of glycine on diamond surface, Eglycine and Ediamond represent the energies of the isolated glycine molecule, and the reconstructed diamond surface structure, respectively.
As the simplest amino acid, glycine (NH2CH2COOH) is made up of a carboxylic group and an amino group, and its functional group is replaced by a hydrogen atom. Figure 1(a) presents its optimized structure, in which the oxygen atom with a single bond to a carbon atom is denoted by O1 and another with double bonds denoted by O2.
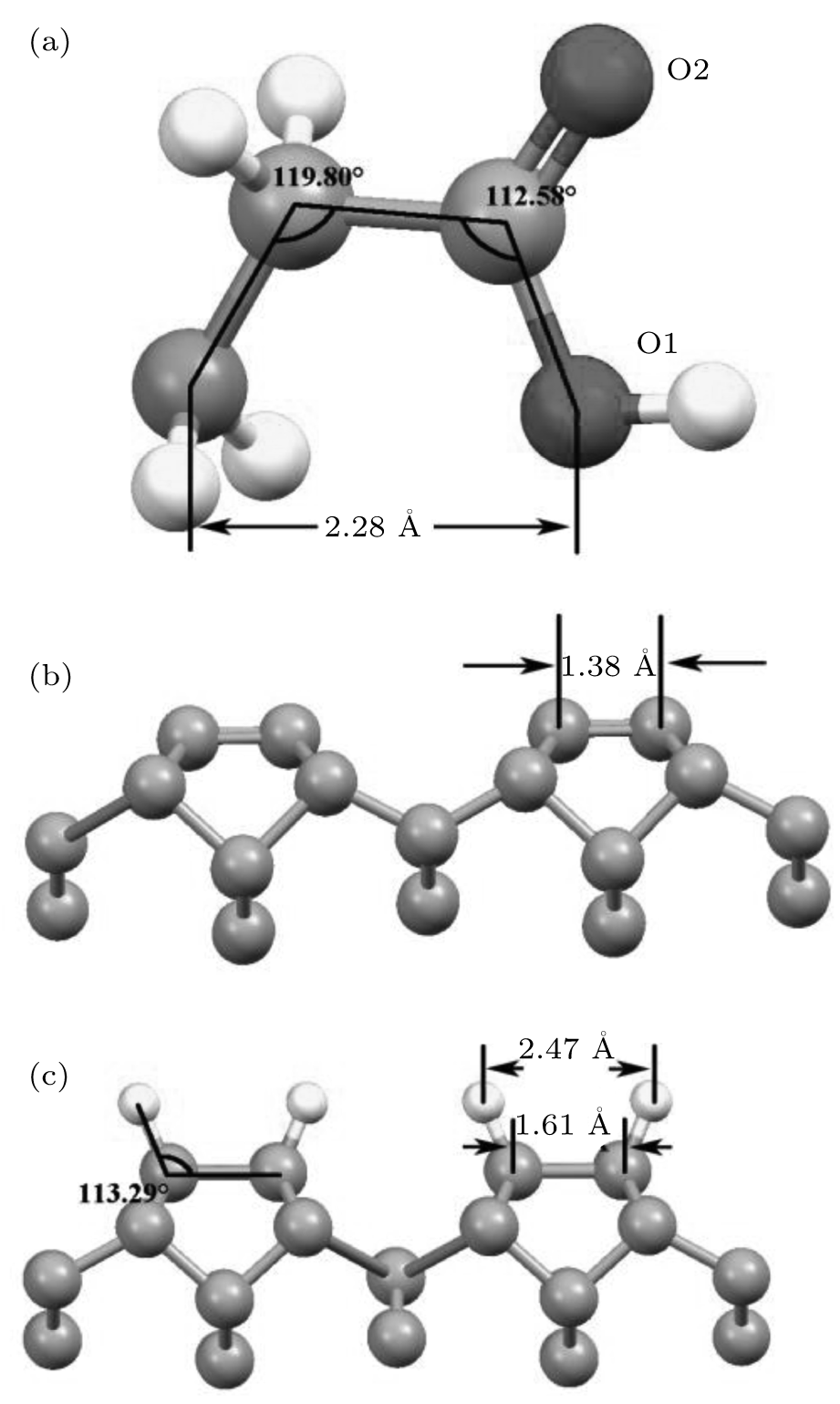 | Fig. 1. (a) The geometry of glycine molecule, C atoms are indicated in grey, and N atoms in blue, O atoms in red, and H atoms in yellow, respectively. (b) Reconstructed diamond (001) 2 × 1 surface. (c) Hydrogen-terminated diamond (001) surface. |
Before the adsorptions of glycine on diamond (001), we primarily optimized the diamond (001) bare surface. During the optimization, the atoms of diamond (001) surface relaxed and dangling bonds of adjacent carbon atoms tied together forming a dimer with a π -bond, as shown in Fig. 1(b). This π -bond is easy to break to interact with adsorbs. The simplest case is that those dangling bonds are passivated by hydrogen atoms as shown in Fig. 1(c), where the H atom is not on the top position of the carbon atom but with a bond angle of 113.29° .[19, 20] This demonstrates the effect of the carbon bond angle during the interaction. This property would greatly influence the interaction with adsorbs, especially for complex molecules.
For glycine molecule there are three active sites, in which two of them are on the carboxylic group (two oxygen atoms) and one on the amino group (a nitrogen atom), respectively. Therefore, when glycine molecule adsorbs to diamond (001), the hydrogen atoms on active sites easily break away from glycine, also the double bond C= O cracks, and the π -bonds of dimers on the reconstructed diamond (001) surface should be opened.
In order to obtain the stable adsorption configurations, our research obeys the rule: all bonds should be saturated upon adsorption, which means that there is no dangling bond existing both on the diamond (001) and the glycine molecule. Following this rule, six stable adsorption structures were found with two dimers opening on the diamond (001) surface. We will discuss these structures in detail based on the number of opened dimers as listed in Table 1.
| Table 1. Number of opened dimers, corresponding figures, adsorption sites, adsorption energy, and deviation of bond angles for glycine on diamond (001). |
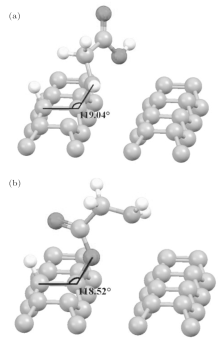
 | Fig. 2. The adsorption structures of glycine on diamond (001) reported in Ref. [14] with bonding sites: (a) NH and (b) OH, respectively. |
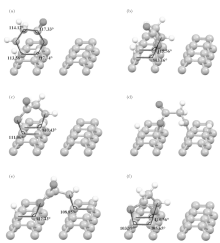
 | Fig. 3. The new adsorption structures for two opened dimers on diamond (001): glycine interacts with carbon atoms (a) at an opened dimer; (b) at the same sides of two adjoining dimers in one row; (c) at different sides of two adjoining dimers in one row; (d) on the trough between dimer rows; (e) on the tough between two staggered dimers; and (f) in a tridentate fashion, respectively. |
We first study the adsorption structures with the one C– C dimer opening on the diamond (001) surface. There are two stable adsorptions reported in Ref. [14], as shown in Figs. 2(a) and 2(b). Figure 2(a) shows nitrogen adsorption and figure 2(b) shows oxygen adsorption with adsorption energies of 2.91 eV and 2.55 eV, and bond angles of 119.04° and 118.52° , respectively.
For two C– C dimers opening on the diamond (001) surface for adsorption, more than one active site in glycine is expected to participate in. We investigated all possible structures and found six stable configurations as shown in Fig. 3, in which five structures are glycine adsorbed in a side fashion and one in a trident fashion.
Figures 3(a)– 3(e) show the side adsorptions of glycine onto diamond (001), where a nitrogen atom and an oxygen atom (O1) in glycine are bonded with the opened dimers in a side fashion, while two hydrogen atoms drop from nitrogen and oxygen to passivate the other dangling bonds of opened dimers. Totally, there are five such configurations: glycine adsorbed on carbon atoms of an opened dimer in Fig. 3(a); on carbon atoms at the same sides of two adjoining dimers in one column in Fig. 3(b); on carbon atoms at different sides of two adjoining dimers in one column in Fig. 3(c); on adjacent carbon atoms belonging to neighboring columns in Fig. 3(d); and on carbon atoms of two staggered dimers in neighboring columns in Fig. 3(e), respectively.
Figure 3(a) shows that glycine is adsorbed just on an opened dimer while two hydrogen atoms dropped from carboxylic acid and amino groups passivate a neighboring opened dimer. The adsorption structure shows that the C bond to N has a bond angle of 113.58° and C bond to O1 117.74° . Both angle values are near to that of free adsorptions on surface, also close to the angle values within glycine molecule, showing a benzene-like hexagonal geometry. The adsorption energy of this structure is as high as 5.11 eV. This value is indeed much larger than the thermal energy kBT (∼ 0.025 eV at 300 K), showing the high stability at room temperature.
In the adsorption structure of Fig. 3(b), glycine molecule in Fig. 3(a) is rotated by 90° to adsorb on surface. Glycine interacts with carbon atoms in the same side of adjoining dimer in the same column and the dropped hydrogen atoms passivate the rest dangling bonds. The bond angles of C– N and C– O are 115.56° and 113.16° , respectively, also close to that of free adsorptions on surface. The adsorption energy of this configuration is 4.77 eV.
Figure 3(c) shows another adsorption structure that the carboxylic and amino groups bonds with carbon atoms at different sides of adjoining dimers in the same column. The bond angles of C– N and C– O are 110.43° and 111.06° , respectively. This adsorption energy is 4.33 eV, and smaller than the former due to a little larger distortions of bond angles and molecules upon adsorption.
Besides the adsorptions on the same dimer row, the nitrogen and oxygen atoms can also interact with carbon atoms belonging to neighboring dimer rows. The trough between dimer rows is relatively larger. However, when the carbon bond angle is taken into account, glycine can be adsorbed as well.
There are two stable adsorption structures on the trough. One of the adsorption structures is shown in Fig. 3(d) with molecule across the neighboring dimer rows. The adsorption energy is 4.48 eV, and the bond angles of C– O and C– N are 110.06° and 110.42° , respectively. Another adsorption structure is shown in Fig. 3(e), the molecule interacts with carbons on two staggered dimers of the neighboring dimer rows. The adsorption energy is estimated to be 4.47 eV and the bond angles of C– O and C– N are 112.43° and 108.85° , respectively.
In above five adsorptions (Figs. 3(a)– 3(e)), only two active sites of glycine interact with the surface. Finally, we consider all three active sites of glycine molecule bond to diamond (001) in a tridentate fashion where two O atoms and one N atom participate in the interactions, similar to that on the Cu (001) surface.[8, 9] The adsorption structure is shown in Fig. 3(f), where two O atoms of glycine bond to one dimer, and a N atom bonds to another adjoining dimer in the same row, and two H atoms dropped from carboxylic and amino sites passivate two dangling bonds of C= O bond and an open dimer respectively. Upon adsorption, the bond angles of C– N, C– O1, and C– O2 are significantly decreased to 110.56° , 105.65° , and 103.59° , respectively. Also the O– O length of glycine is increased from 2.28 Å to 2.31 Å due to the carbon bond angle. This larger distortion will cost much energy. Thus, the adsorption energy in this configuration is 4.29 eV, which is indeed smaller than that in (Figs. 3(a)– 3(e)) adsorption structures with only one O atom and one N atom in side fashion.
The above six structures adsorbed on two opened dimers on the surface all have large adsorption energies, much more stable than the adsorptions on one dimer shown in Fig. 2. Thus, all those structures may be regarded to be stable and may coexist. However, the best adsorption structure has the closest geometry to benzene-like hexagonal geometry. Note that different to the adsorption on a metal surface, glycine prefers to be adsorbed in a side fashion with only O1 atom and N atom on the diamond (001) surface.
It is worthwhile to analyze the above adsorption structures in detail. Before calculations, it might be expected that the structure in trident fashion in Fig. 3(f) would have the largest adsorption energy because all three active sites participate in the interaction similar to that on metal surfaces, and also expected that the structure in Fig. 3(a) would have the weakest adsorption because of the large length difference between carbon dimer length 1.61 Å and N– O1 length of glycine 2.77 Å . However, just the opposite to our expectation, figure 3(a) is the best adsorption structure while figure 3(f) is the worst among these six structures. Both are attributed to the same physical origin that is the carbon covalent bond angle. One can see that glycine in Fig. 3(a) is the most comfortably accommodated on an opened dimer with bond angles only slightly differing to the free adsorption configurations in Figs. 1(a) and 1(b). However, adsorption in trident form in Fig. 3(f) is greatly distorted where the bond angles are largely decreased and the O– O distance in glycine increases from 2.28 Å to 2.31 Å . The distortion of bond angles and molecule upon adsorption will cost energy and lead to a high energy adsorption structure. Therefore, the importance of carbon bond angle is embodied in all adsorption structures. The best adsorption structure is the one that has the largest adsorption energy with the smallest distortion of carbon bond angles shown in Fig. 3(a). Such peculiar behavior of carbon atoms has also presented on the step diamond (001) surface[21] with short and strong bond properties.
Furthermore, from obtained bond angle values of all adsorption structures on the surface, we quantitatively analyzed the bond angle deviations by calculating mean square displacement to standard values of the “ free” adsorption structures in Figs. 2(a) and 2(b). The results are listed in the last column of Table 1. The analysis is consistent with the adsorption results that the smallest deviation has the largest adsorption energy and vice versa.
In this work, we have systematically investigated the adsorption properties of glycine on diamond (001) surface with first principles calculations and considered all possible configurations without any dangling bond on the surface and on the glycine molecule upon adsorption. It is found that two dimers opening adsorptions on diamond (001) are much better than only one dimer opening adsorptions reported in the literature. The best adsorption structure has the benzene-like hexagonal geometry with the most comfortable accommodation on an opened dimer. It is emphasized that the carbon covalent bond angle plays a crucial role during adsorption on the diamond (001) surface. This work gives a comprehensive understanding of adsorption of glycine on diamond (001), and provides useful information for the development of high-performance diamond biosensors.
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|