




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Structural, elastic, and electronic properties of sodium atoms encapsulated type-I silicon–clathrate compound under high pressure
[Zhang Wei†a), d)  , Chen Qing-Yun
, Chen Qing-Yunb) , Zeng Zhao-Yic) , Cai Ling-Cangd) ]
, Chen Qing-Yun|
|


†Corresponding author. E-mail: zwphys@gmail.com
*Project supported by National Natural Science Foundation of China (Grant Nos. 11347134 and 11304254) and the Doctor Foundation of Southwest University of Science and Technology, China (Grant No. 13zx7125).
We calculated the structural, elastic, and electronic properties of alkali metal Na atoms doped type-I silicon–clathrate compound (Na8Si46) under pressure using first-principles methods. The obtained dependencies of bond lengths and bond angles on pressure show heterogeneous behaviors which may bring out a structural transition. By using the elastic stability criteria from the calculated elastic constants, we confirm that the Na8Si46 is elastically unstable under high pressure. Some of the mechanical and thermal quantities include bulk modulus, shear modulus, Young’s modulus, Debye temperature, sound velocity, melting point, and hardness, which are also derived from the elastic constants. The calculated total and partial electron densities of states of Na8Si46 indicate a weak interaction between the encapsulated Na atoms and the silicon framework. Moreover, the effect of pressure on its electronic structure is also investigated, which suggests that pressure is not a good choice to enhance the thermoelectricity performance of Na8Si46.
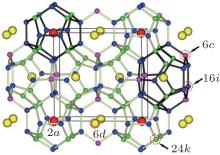
The lattices of type-I silicon– clathrate are cubic and are formed from eight face-shared polyhedral silicon cages, as illustrated in Fig. 1. These cages are bridged by direct covalent bonding between the Si atoms. In this cubic unit cell, there are two small pentagonal dodecahedron Si20 cages and six large tetrakaidecahedron Si24 cages, each cage contains one sodium atom at its center, the Wyckoff symmetry sites of these two types of sodium atoms are 2a and 6d sites, respectively. The unique Si atoms locate at three distinct Wyckoff positions, i.e., 6c, 16i, and 24k, as labelled in Fig. 1. In the crystallographic description using the Wyckoff position, the number indicates the multiplicity of Wyckoff sites, which tells us how many atoms of one type there are in the unit cell while the letter is simply a label and has no physical meaning. In this way, we can distinguish and count all of the different types of interatomic distances and bond angles. However, for convenience, in the following description we use numbers 1, 2, and 3 to represent the sites of 6c, 16i, and 24k, while numbers 4 and 5 represent the 2a and 6d sites, respectively. Silicon– calthrate compounds were first synthesized by Cros et al. early in 1965; [1, 2] however, they came back to attention only recently following the discovery of superconductivity in both Ba and Na atoms intercalated clathrate BaxNaySi46.[3] They are currently attracting increasing attention because of their potential applications in areas of superconductivity, [4] large gap semiconductors, [5] optoelectronics, [6] and thermoelectrics, [7, 8] etc.
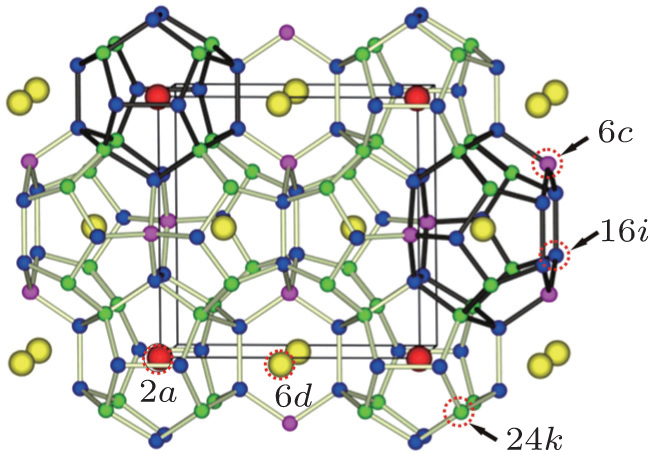 | Fig. 1. The sketch map of the crystal structure of Na8Si46. |
To study the properties of alkali metal Na atoms, intercalated silicon– clathrate under high pressure is motivated by both fundamental and applied reasons. The main applications of such clathrate compounds are due to the particular interaction between guest atoms and the host lattice, the utilization of pressure can be an effective method to explore the mechanism of this interaction, which is very important to understand most of the clathrate properties. In addition, to synthesize such high quality compounds is difficult in experiments. The most well known method to synthesize such kinds of silicon– clathrates uses zintl compounds as precursors and through different types of chemical reactions under deep vacuum or at ambient pressure.[1, 9] Since the first high pressure synthesis of type-I Ba8Si46 clathrate was realized in 2000 by Yamanaka et al.[10] in a multi-anvil press, increasing numbers of analogues have been synthesized using high pressure techniques.[11, 12] Only recently, Na– Si clathrates have been simultaneously obtained for the first time by two groups using high pressure techniques.[13, 14] However, as declared in Ref. [13], a well-controlled Na8Si46 crystal growth needs more detailed knowledge about its properties under high pressure, which is still wanting. Although some experiments have been performed to study the high pressure stability of Na8Si46, [15, 16] the highest pressure was only applied to 13 GPa and even the acquired data were insufficient for a fit with Birch– Murnaghan equation of states (EoS) and were unable to give the bulk modulus. In Stefanoski’ s experiments, the transport properties of Na8Si46 such as temperature-dependent resistivity and heat capacity were studied.[17] The theoretical studies of Na8Si46 have mainly focused on the thermodynamic[18] and electronic properties[19, 20] under ambient pressure. However, systemic theoretical studies of the properties of Na8Si46 under high pressure are little-reported. Consequently, in this work we will investigate the structural, elastic, and electronic properties of Na8Si46 under high pressure, which are believed to offer a benefit reference for the application and synthesis of this compound.
We employed the ultrasoft pseudopotentials introduced by Vanderbilt[21] for all of the ion– electron interactions in the electronic structure calculations. Both the local density approximation (LDA)[22] as well as the generalized gradient approximation (GGA)[23] were used to calculate the exchange– correlation energy. During the pseudo-atomic calculations, the valence electron densities were defined by Na (2s, 2p, 3s) and Si (3s, 3p). A plane-wave basis set with energy cut-off 600 eV was applied, which could ensure the self-consistent converge of the total energies achieve 10− 6 eV/atom. The crystal reciprocal-lattice and integrations over the first Brillouin zone have been performed using 6× 6× 6 Monkhorst– Pack. Combined with the variable cell approach, hydrostatic pressure was realized by employing the Parrinello– Rahman method.[24, 25] At each target external pressure, a full optimization of the cell structure has been performed by applying the Broyden, Fletcher, Goldfarb and Shanno (BFGS) scheme.[26] All of these total energy electronic structure calculations were carried out with the Cambridge Serial Total Energy Package (CASTEP) code.[27, 28]
To investigate the mechanical properties of Na8Si46 clathrate compound, the elastic stiffness tensors were calculated, which relate to the stress and the strain tensors by Hooke’ s law. Because of symmetrical characteristics of the stress and strain tensors, most general elastic stiffness tensors only have 21 non-zero independent components. In the present work, Na8Si46 is a cubic crystal, thus these independent components are reduced to three components C11, C12, and C44. After computing the stress generated by forcing a small strain to an optimized unit cell, these second-order elastic stiffness coefficients can be determined by means of linear fitting of the stress– strain curves.[29, 30]
Through both the GGA and LDA schemes in electronic calculations, we obtained the equilibrium structure of Na8Si46. The resulting static lattice constants, interatomic distances, and angles formed by silicon atoms of different sites are listed in Table 1 to Table 3, respectively, together with other available theoretical[31] and experimental results[2, 13, 17, 18] which are provided for comparison. Not surprisingly, it can be found that both the lattice constants and the interatomic distances calculated by GGA are larger than those by LDA due to different approximations. Through a comparison with the experimental results, the calculated results by GGA approximation in the present calculation are somewhat better than those by LDA approximation and the other theoretical results obtained at the HF-LCCO level with 6-21G and 6-21G* basis sets for Si.[31] A comparison with Si46 shows that its lattice constant is 10.08 Å .[32] It is also found that the lattice constant of Na8Si46 has a slight increase, which indicates the intercalation of Na atoms caused a very small expansion of the silicon– clathrate framework. Of course, as the ionic radius of the intercalated metal atom increases, the lattice constants will increase obviously, such as the case of K[33] and Ru[34] atoms intercalation.
| Table 1. The calculated lattice constants (Å ) of Na8Si46 at the theoretical equilibrium volume. |
| Table 2. The interatomic distances of Na8Si46. |
| Table 3. The interatomic angles of Na8Si46. |
In order to investigate the structural properties of such cage-like structures in detail, the bond angles formed by silicon atoms at different sites are calculated and counted, which are given in Table 3. This information can reflect the local structure characteristics of the lattice. It can be found that the theoretical results are consistent with the experimental results except for some angles which have about a one-degree difference.
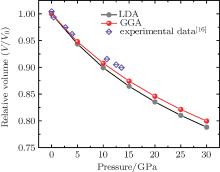
In Fig. 2, we illustrate the dependence of normalized cell volume of Na8Si46 with pressure together with available experiments from San– Miguel.[16] However, the pressure in these experiments was applied to 13 GPa. It can be found that the GGA results show little better results than those of LDA, just like the calculation of lattice constant. Thus, in the following discussion, the pressure dependences of various mechanical properties of GGA results are given. From the equation of state (EoS) we found that when the pressure reaches to 30 GPa, its volume is reduced to about 20%, this compression behavior is very similar to another type-I silicon– clathrate intercalated by alkali metal potassium atoms (K8Si46).[35] However, there was a distinct discontinuity at 20 GPa in the experimental EoS which suggested a structural change of K8Si46. In the case of Na8Si46, this anomalous change in unit cell volume has not been found.
 | Fig. 2. Variation of the relative volume of type-I clathrate Na8Si46 with pressure. |
The evolutions of the Si– Si and Na– Si interatomic distances with pressure are displayed in Figs. 3 and 4. In Fig. 3 it can be found that the dependences of Si– Si interatomic distances on pressure show different tendencies. At the framework of the silicon cage, the variety of interatomic distances of Si(3)– Si(3) with pressure has a turning point at 10 GPa, corresponding to two different compress characteristics under pressure. The compression behavior of Si(3)– Si(1) and Si(2)– Si(3) are very similar, and the bond Si(2)– Si(2) is the most difficult one to be compressed. The experimentally measured average distance of Si– Si atoms is also given in Fig. 3 for comparison.[16] As shown in Fig. 4, it can be seen that the pressure dependence of Na– Si interatomic distances are in general the same and parallel to each other, except for Na(4)– Si(3) which presents a more slow variety under high pressure.
 | Fig. 3. Calculated pressure dependencies of interatomic distance of silicon atoms at the Na8Si46 clathrate framework. |
 | Fig. 4. Calculated pressure dependencies of interatomic distance between silicon atoms and the host Na atoms. |
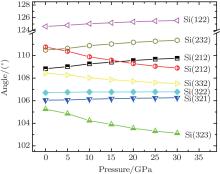
Figure 5 illustrates the variations of angles formed by silicon atoms at the framework of clathrate with pressure. It is found that during all of the compression processes, the angles Si(3– 2– 2) and Si(3– 2– 1) are almost constant; however, other angles display different trends with the increase of pressure. The angles Si(1– 2– 2) and Si(2– 3– 2) increase with the enhanced pressure while angles Si(3– 3– 2) and Si(3– 2– 3) decrease with the increasing pressure. It is noted that there are two types of angle Si(2– 1– 2) that present an opposite variety with pressure and they have a cross point at about 17 GPa. The different dependences of local structure of the clathrate on pressure may lead a structural transition, which will be confirmed in the following discussion of the elastic properties.
 | Fig. 5. Calculated variations of angles formed by silicon atoms at different sites of Na8Si46 clathrate with pressure. |
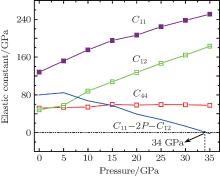
The calculated elastic constants of Na8Si46 under zero pressure are listed in Table 4. For the Na8Si46 compound, there are three independent elastic constants; i.e., C11, C12, and C44. It is found that the calculated C11 which represents the uniaxial deformation along the [0 0 1] direction in crystal is the largest. We know that C11 relates to the melt property of the crystal and an empirical relation proposed by Fine et al.[36] can be expressed by Tm = 553 K + (591/1011 Pa)C11 ± 300 K, the obtained melting temperature of Na8Si46 is 1310.7 K (GGA) and 1377.4 K (LDA) plus or minus 300 K. By Monte Carlo simulations, Miranda et al. determined the melting point of type-I Si46 and type-II Si34 clathrate structures to be (1482± 25) K and (1522± 25) K, respectively.[37] The experimental melting point value of Si34 was found to be 1473 K.[38] Regardless of the estimation error, it seems that the melting point of Na8Si46 is reduced by the intercalation of Na atoms. Moreover, in the cubic crystal, if the calculated C12 equals C44, then the interatomic force might be central.[39] It is noted that in the present work both GGA and LDA approximations give very close values of C12 and C44, which indicates the anisotropic interactions between two atoms in the Na8Si46 crystal.
| Table 4. The calculated zero pressure elastic constant Cij, bulk module B, shear modulus G, Young’ s modulus E, E(100), E(111) (in unit GPa), Poisson’ s ratio σ , the anisotropy factor A, and the Pugh’ s ratio k. |
Through the calculated elastic constants, some of the physical quantities related to the mechanical and thermal properties of the material can be obtained. For a cubic structure Na8Si46, the bulk modulus B, the shear modulus G, and anisotropy factor A are given by
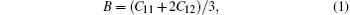

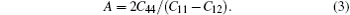
The Young’ s modulus E, the Poisson’ s ratio σ , and Pugh’ s ratio k are then taken as
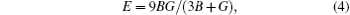
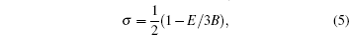

Moreover, the Young’ s modulus E along [100] and [111] directions in crystal can be obtained by the following relations


The transverse and longitudinal sound velocities vs and vl can be derived from Navier’ s equation, as follows:[40]
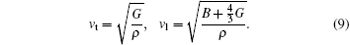
From the average sound velocity vm, Debye temperature Θ D is estimated by[41]

where h is Planck’ s constants, kB is Boltzmann’ s constant, NA is Avogadro’ s number, n is the number of atoms per formula unit, M is the molecular mass per formula unit, ρ is the density, and Vm is calculated from[41]

The calculated bulk module B, shear modulus G, Young’ s modulus E, E(100), E(111), Poisson’ s ratio, anisotropy factor A, and Pugh’ s ratio k of Na8Si46 are given in Table 4. It is noted that the bulk modulus calculated in the present work is smaller than the silicon diamond structure, which is 97.88 GPa.[42] However, in the case of other metal atoms intercalation, [42] San– Miguel et al. suggested that the guest– host hybridization could enhance the B values higher than those of the empty clathrates and are even close to the values of their diamond structure. This happens because the hybridization between Na atoms and the silicon framework is very weak, which can be seen clearly in the following electronic density of state calculations. The pity is that the acquired experimental data are too few to give the bulk modulus by a fit of the Birch– Murnaghan equation of states. The difference between Young’ s modulus along [100] and [111] crystallographic directions is about 20%, this indicates that the Na8Si46 crystal is elastically anisotropic, this conclusion is confirmed by anisotropy factor A because it is larger than 1. The Poission’ s ratio σ and Pugh’ s ratio k are closely correlated to the brittle and ductile behavior of materials. According to Frantsevich’ s rule, [43] if the Poisson ratio is less than 1/3, then the materials will present brittle characteristics and if this value is larger than 1/3 then they can be regarded as ductile materials. The Poisson’ s ratio calculated here indicates that the Na8Si46 crystal will have a brittle behavior. Analogous to σ , the Pugh’ s ratio k = 0.57 shows the borderline of the ductile and brittle properties of the materials, a lower k value means that the materials are ductile, otherwise they are brittle.[44] The obtained conclusion from Pugh’ s ratio in the present work is the same for the Poisson’ s ratio.
In general, the hardness of the clathrate compounds is governed by the the framework forming atoms. By a liner fit of the dependence of Vickers hardness on the shear module and Young’ s module of dozens of type-I silicon-based intermetallic clathrates, the two empirical relations were given as: Hv = 0.058E and Hv = 0.141G.[45] The obtained hardness values are almost identical, equal to 6.73 GPa and 6.60 GPa, respectively. It can be found that this hardness value is similar to melanophlogite [Si46O92], which is a silicate analog of the type-I silicon– clathrate with the values of Vickers hardness between 6.5 GPa and 7 GPa.[46] It is noted that their hardness is just comparable to quartz, for which the Vickers hardness is 7 GPa.[46] However, the hardness of another type-I silicon– clathrate with iodine atoms intercalated (I8Si46) is close to its silicon diamond structure.[47] The difference may originate from the stronger hybridizations between the iodine and Si network compared to Na-doped clathrate.
Besides hardness, the sound velocity can also be obtained from the shear modulus and bulk modulus, and Debye temperature can thus be deduced from the value of sound velocity. In Table 5, the only available experimental result of the Debye temperature estimated by employing the experimental atomic displacement parameters and Debye model is given.[48] The good agreement between it and our calculation suggests the validity of elastic constants obtained in the present calculation. In this work, the sound velocities of both longitudinal and transverse waves along three different crystallographic directions including [100], [110], and [111] are calculated, the formulas used in sound velocities calculations are given by
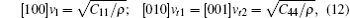
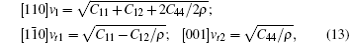
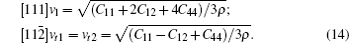
| Table 5. The calculated zero pressure sound velocity (km/s) and Debye temperature (K). |
From Table 5, it can be seen that the cubic Na8Si46 compound has large sound velocities due to its small density and large elastic constants Cij. The difference of calculated sound velocities along different directions also shows the anisotropy of Na8Si46 crystal, just like Young’ s modulus.
In order to investigate the high pressure stability of Na8Si46, the elastic constants under high pressure are also calculated, which are displayed in Fig. 6. It can be found that both the C11 and C12 increase with the enhanced pressure while C44 is barely changed. It is known that the mechanical stability under isotropic pressure for a cubic crystal can be judged from the following criterion[49]

where 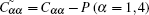



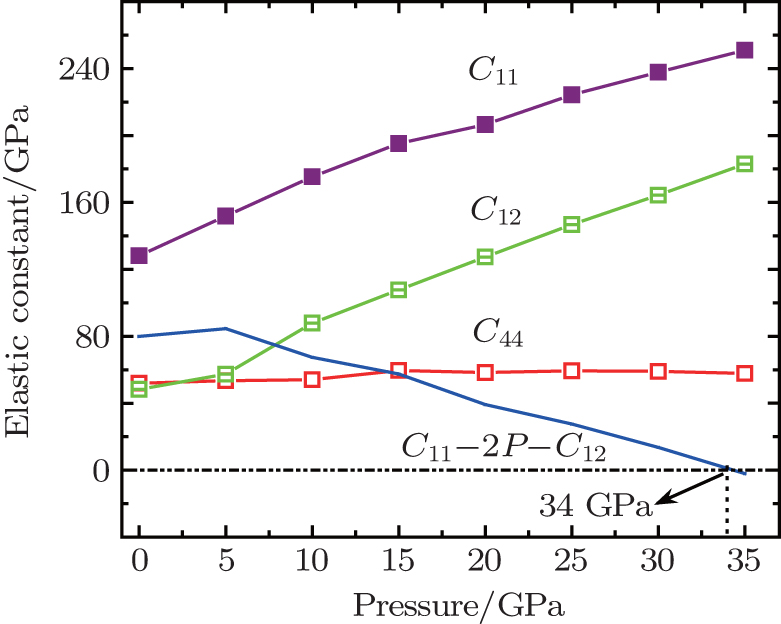 | Fig. 6. The pressure dependence of elastic constants of Na8Si46. |
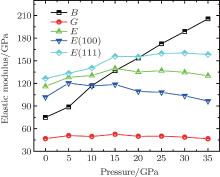
Moreover, some other pressure dependencies of elastic modulus are also calculated, as shown in Fig. 7. It is found that bulk modulus increases with the enhanced pressure while shear modulus is not sensitive to the change of pressure. As the pressure increases, the Young’ s modulus first increases and then decreases. The difference of Young’ s modulus along crystallographic directions [100] and [111] becomes increasingly larger, indicating an intensification of crystal anisotropy. This coincides with the enlargement of the anisotropy factorA, which is 1.7 when the pressure is applied to 35 GPa. Furthermore, when the pressure is enhanced to 35 GPa, the Poisson’ s ratio will increase to 0.42 and Pugh’ s ratio decreases to 0.23, which indicates an obvious ductile behavior of Na8Si46 under high pressure. The dependence of the transverse and longitudinal waves along three directions are also calculated. It is found that the spread speed of longitudinal waves increase with the enhancement of pressure while transverse wave speed decreases as pressure increases. The average sound speed presents a behavior that is just like the transverse waves.
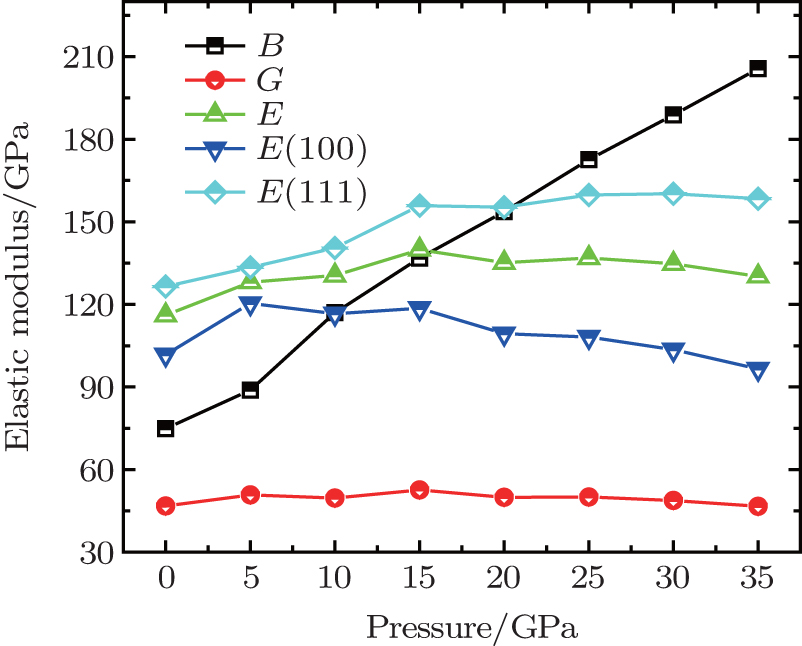 | Fig. 7. The pressure dependence of bulk module, shear modulus, and Young’ s modulus of Na8Si46. |
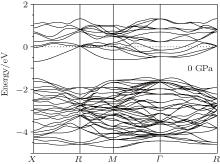
Figure 8 shows the band structures of Na8Si46 clathrate along the symmetry line of the simple cubic Brillouin zone in the region near the Fermi level. It can be seen that the Fermi level lying in the conduction band indicates the metallic behavior of the Na8Si46 clathrate compound. By using the linearized augmented plane wave (LAPW) method, the band structure of Si46 given by Kurganski et al. was almost identical to the present calculated band structure of Na8Si46.[19] This phenomenon is not surprising and it can be interpreted using the rigid-band model.[50] According to this model, isostructural compounds have similar energy band structures, such as Si46 and Na8Si46 clathrates. However, after the intercalation of Na atoms in eight empty silicon cages, the conduction-band edge will be formed by the contribution from eight valence electrons of Na atoms, which results in a shift of the Fermi level to higher energies, and thus the Fermi level is located into the conduction band.
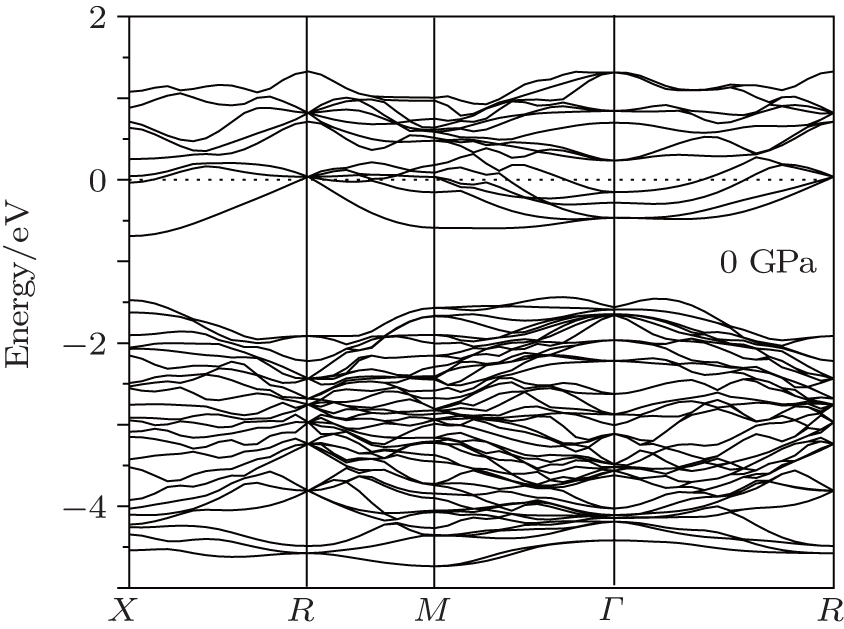 | Fig. 8. The band structure of Na8Si46 under zero pressure. |
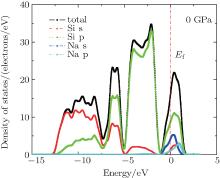
The total and partial density of states (DOS) are given in Fig. 9, from which we find that the contribution of silicon p states is predominant near the valence band top and that of s states is predominant at the bottom sides of valence band. The conduction bands are mainly comprised of silicon p states and Na s states. However, the contribution made by the states of Na atoms to the total DOS in the conduction band is less than a half of the contribution from Si states, so the conduction band density of states is not modified strongly upon the inclusion of Na atoms in comparison with the pristine Si46, which confirms a weak hybridization between the Si46 conduction-band states and Na states. Unlike the case of Ba8Si46 and I8Si46 clathrates, the conduction bands are strongly modified by the Ba and I states.[20, 48]
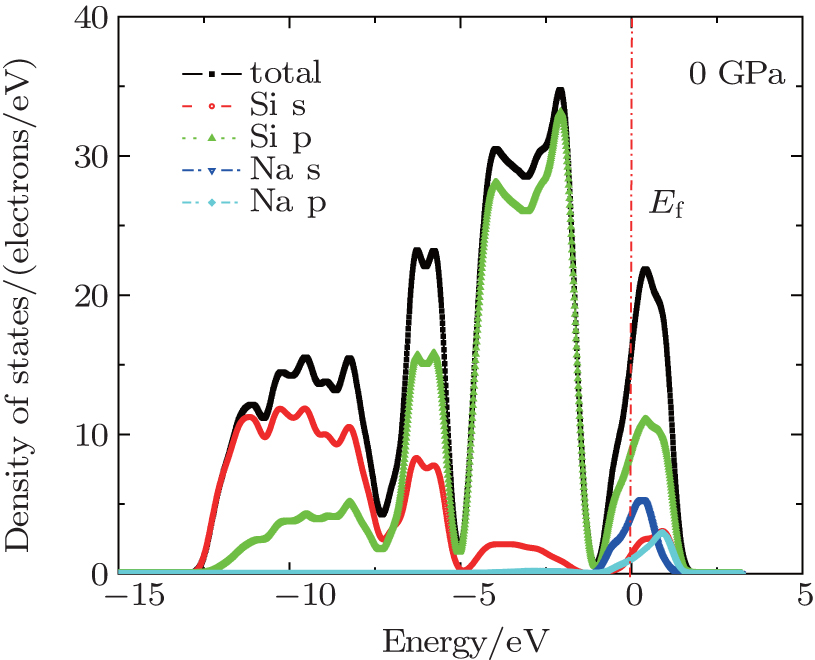 | Fig. 9. The electronic density of states of Na8Si46 under zero pressure. |
Pressure usually has a strong effect on the electronic properties of materials, the band structure and DOS of Na8Si46 under 25 GPa are shown in Figs. 10 and 11, respectively. It is found that pressure can cause an energy-gap narrowing between conduction band and valence band while the width of bands at the bottom of the conduction band become more wider. This indicates that the electrons have more freedom in this energy range. The Fermi-level density of states N(EF) of Na8Si46 calculated under zero pressure is 16.3 states· eV− 1· f.u.− 1. When the pressure is increased to 25 GPa, this value decreases to 11.6 states· eV− 1· f.u.− 1. This finding suggests that the pressure can reduce the electrical conductivity of this clathrate compound, so pressure may not be a suitable choice to enhance the thermoelectric performance of Na8Si46.
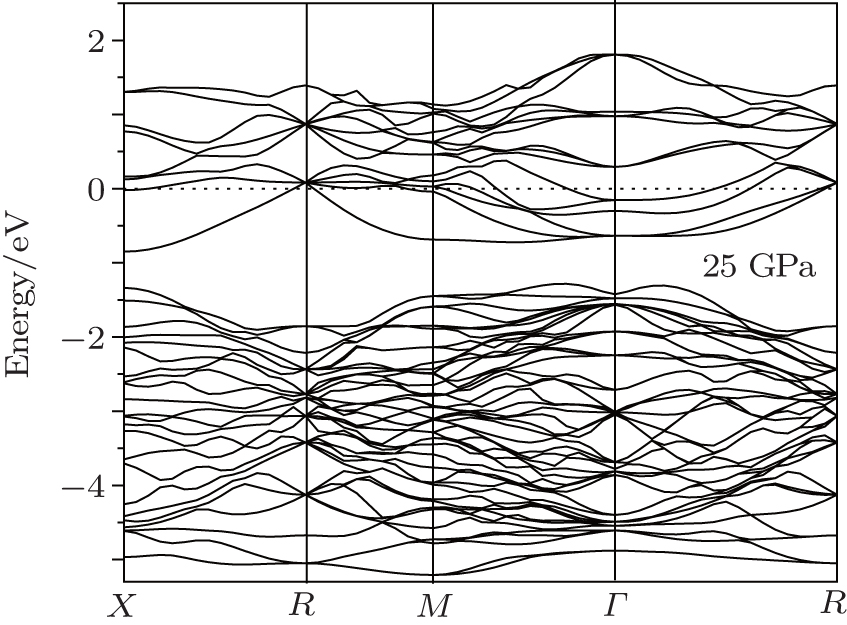 | Fig. 10. The band structure of Na8Si46 under 25 GPa. |
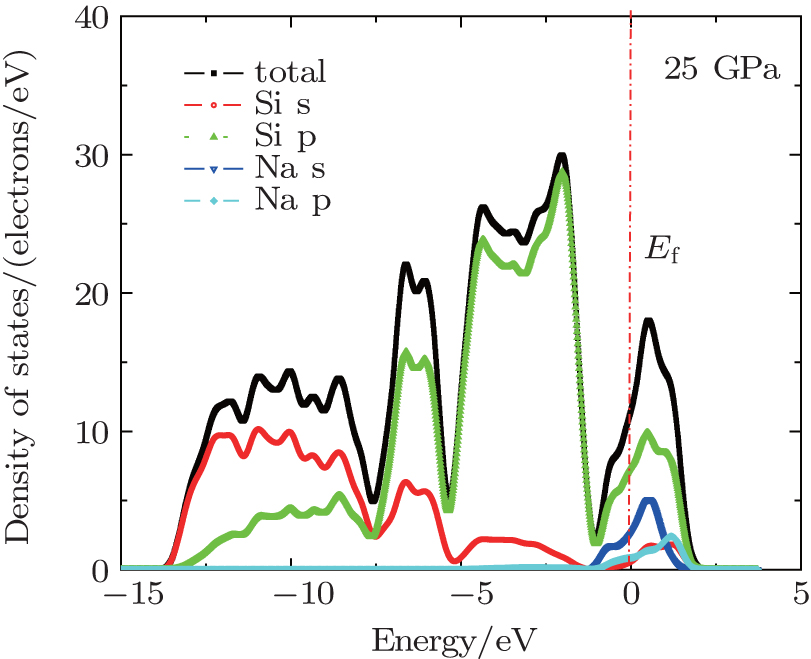 | Fig. 11. The electronic density of states of Na8Si46 under 25 GPa. |
In summary, we have given a detailed study of the structural, elastic, and electronic properties of Na8Si46 under high pressures. The local structure evolutions with pressure show different behaviors, which may be responsible for the structural phase transition under high pressures. The mechanical stability criterion confirms that this compound is not stable under high pressure. Moreover, through the calculated elastic constants, some of the mechanical and thermal quantities are derived. The Young’ s modulus and sound velocities along different crystallographic directions having different values reveal the anisotropy of Na8Si46 crystal and the pressure is found to make an intensification of the anisotropy. The calculated electronic band structure presents a metallic character of Na8Si46. By analyzing the partial density of states of Na8Si46, we find a weak interaction between the doped Na atoms in the cage and the silicon atoms located at the framework. As the pressure increases, the Fermi-level density of states decreases, which will have a negative effect in the application of thermoelectricity.
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|
| 35 |
|
| 36 |
|
| 37 |
|
| 38 |
|
| 39 |
|
| 40 |
|
| 41 |
|
| 42 |
|
| 43 |
|
| 44 |
|
| 45 |
|
| 46 |
|
| 47 |
|
| 48 |
|
| 49 |
|
| 50 |
|