



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
New crystal structure and physical properties of TcB from first-principles calculations
[Zhang Gang-Tai†a), b)  , Bai Ting-Ting
, Bai Ting-Tingc) , Yan Hai-Yan‡d) , Zhao Ya-Rua) ]
, Bai Ting-Ting, Zhao Ya-Ru|
|


†Corresponding author. E-mail: gtzhang79@163.com
‡Corresponding author. E-mail: hyyan1102@163.com
*Project supported by the Science Foundation of Baoji University of Arts and Sciences of China (Grant No. ZK11061) and the Natural Science Foundation of the Education Committee of Shaanxi Province, China (Grant Nos. 2013JK0637, 2013JK0638, and 2014JK1044).
By combining first-principles calculations with the particle swarm optimization algorithm, we predicted a hexagonal
Designing and searching for ultra-incompressible and superhard materials are of great scientific interests owing to their various industrial applications, such as cutting and polishing tools, abrasives, oil exploitations, and coatings. Generally, there are two kinds of materials that are regarded as potential candidates for superhard materials. One is the strong covalent-bonded compounds formed by light atoms, such as diamond, [1]c-BN, [2] B6O, [3] BC2N, [4] and BC5.[5] The other is by combining heavy transition metal (TM) atoms with light and covalent bonding atoms, such as OsB2, [6] ReB2, [7] RuC, [8] ReN4, [9] OsO2, [10] etc. The compounds formed by transition metal and light atoms usually possess high valence electron density and directional covalent bonds, and these covalent bonds are strong enough to inhibit creation and movement of dislocations, which significantly improve their mechanical properities and create high hardness. In view of this point, recent design of the new intrinsically potential superhard materials has concentrated on light element TM compounds. A primary example and among the first synthesized materials following this principle is OsB2, [6] which is expected to be a superhard material (Hv ≥ 40 GPa). However, further calculations and experiments reveal that it is only a hard material.[11– 13] Later, Yong et al.[14] synthesized OsN2 at high pressures and temperatures, and the obtained zero-pressure bulk modulus was comparable with those of the traditional superhard materials. Many years earlier, Kempter and Nadler[15] claimed that they have synthesized WC-type OsC at ambient pressure and high temperature. No other experimental synthesis of osmium carbide has been reported since then. Recently, Guo et al.[16] investigated the structure and mechanical properties of OsC with nine structures and proposed that the synthesized OsC should be in NiAs structure. Liang et al.[17] systematically studied the electronic structure and mechanical properties of OsB, OsC, and OsN in the WC, NaCl, CsCl, and ZnS structures and also found that only four phases are mechanically stable but none of them is superhard. Very recently, Zhang et al.[18] reported an orthorhombic Pmmn structure for OsB4 with high bulk modulus (294 GPa) and large hardness (28 GPa) energetically much superior to the previously reported WB4-type structure.
Technetium (Tc) lies to the top left of Os in the periodic table, which should have a low compressibility, so it is worth studying the mechanical properties of its borides, carbides, and nitrides. Theoretically, technetium-based materials have been extensively studied.[19– 25] For example, Wang[19] proposed the hexagonal structure of TcB2 is more stable than the orthorhombic one and showed it is a potential superhard material. Liang et al.[20] reported that TcC and TcN in two hexagonal phases are ultra-incompressibile. Aydin and Simsek[21] investigated the structure, mechanical, and electronic properities of TcB2 and also showed that TcB2 within ReB2-type is energetically favorable than that of the AlB2-type one and it is a hard material. Deligoz et al.[22] investigated the lattice dynamical and thermodynamical properties for TcB2 in AlB2, ReB2, and OsB2-type structures. Zhong et al.[23] studied the phase stability, physical properties, and hardness of TcB2 in the ReB2 and OsB2 structures. Wu et al.[24] established the phase stability of technetium borides with various stoichiometries and also showed that the predicted Cmcm structure is more stable than the previously proposed WC-type structure.[25]
To the best of our knowledge, technetium monoboride (TcB) has not been experimentally synthesized so far due to the technical difficulty, besides, TMBs can be synthesized under ambient pressure, which leads to the low-cost synthesis condition and is beneficial to their applications. Because of this, in this paper we shall extensively investigate the groundstate structure of TcB by the ab initio particle swarm optimization (PSO) approach for crystal structure prediction.[26] This method has been successful in predicting crystal structures for various systems, [27– 29] unbiased by any known information. A hexagonal 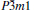
The PSO methodology for crystal structural prediction has been implemented in the crystal structure analysis by the particle swarm optimization (CALYPSO) code[31] with 1∼ 4 formula units (f.u.) per simulation cell. The underlying calculations are performed using density functional theory within the generalized gradient approximation (GGA), as implemented in the Vienna ab initio simulation package (VASP).[32– 34] The electron and core interactions are included by using the frozen-core all-electron projector augmented wave (PAW) method, [35] where the 2s22p1 and 4p64d55s2 are considered as valence electrons for B and Tc, respectively. The cutoff energy 520 eV and proper Monkhorst– Pack k meshes (14 × 14 × 6)[36] are used to ensure that the total energy calculations are well converged to better than 1 meV/atom. The phonon frequency is calculated by using a supercell approach as implemented in the PHONOPY code.[37] Single crystal elastic constants are determined by the strain– stress method. The bulk modulus, shear modulus, Young’ s modulus, and Poisson’ s ratio are derived from the Voigt– Reuss– Hill approximation.[38] The quasi-harmonic Debye model is applied to study the lattice thermal expansion and the specific procedure of calculation can be found elsewhere.[30]
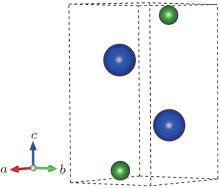
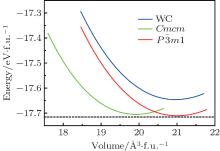
At 0 GPa, a new stable structure with space group 

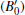
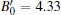

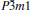
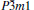
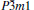
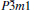
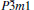
 | Fig. 1. Crystal structure of  |
 | Fig. 2. Total energy versus f.u. volume for three different phases of TcB. |
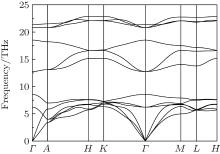
 | Fig. 3. Phonon dispersion curves of 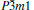 |
The mechanical properties of the 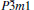

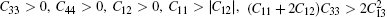
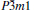

Table 1. Calculated elastic constants Cij (GPa), bulk modulus B (GPa), shear modulus G (GPa), Young’ s modulus E (GPa), Poisson’ s ratio v, and B/G of  |
 | Fig. 4. Calculated volume compressions as a function of pressure for TcB in different phases compared with c-BN, OsB, and RuB. |
The elastic anisotropy of crystals can exert great effects on the properties of the physical mechanism, such as anisotropic plastic deformation, crack behavior, and elastic instability. Therefore, it is important to study the elastic anisotropy to improve their mechanical durability. The shear anisotropic factors provide a measure of the degree of anisotropy in the bonding between atoms in different planes. For hexagonal 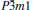
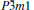
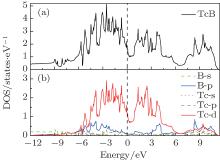
To understand the mechanical properties on a fundamental level, the total and partial densities of states (DOS) are plotted in Fig. 5. Obviously, the adequately large total DOS at the Fermi level indicates that 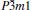
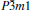
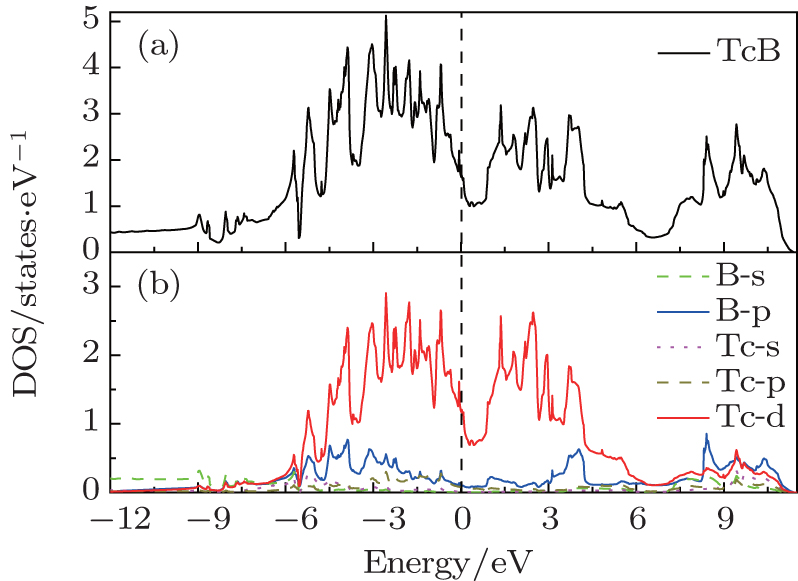 | Fig. 5. Total (a) and partial (B) densities of states (DOS) of  |
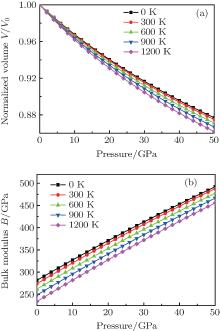
The investigation on the thermodynamic properties of solids at high pressure and high temperature is an interesting topic in materials science. Thus the thermal properties of 

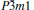
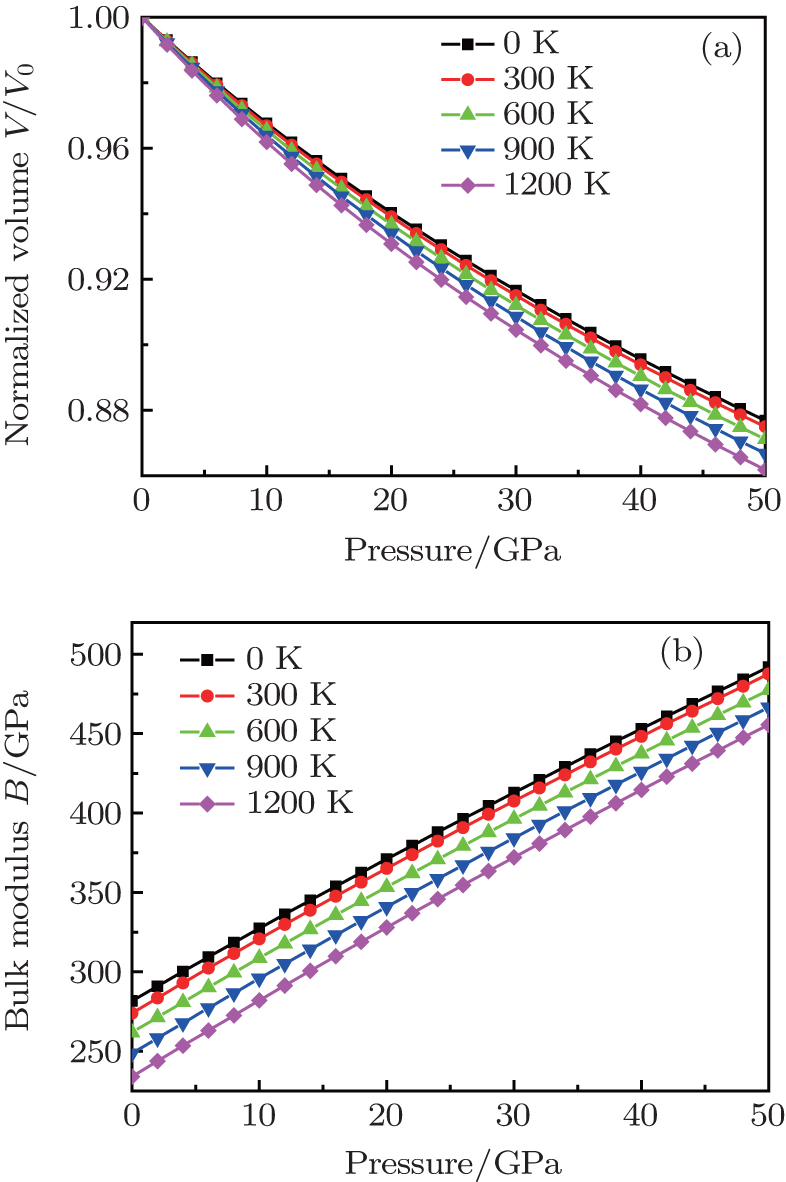 | Fig. 6. Calculated normalized volume V/V0 (a) and bulk modulus B of  |
As two key thermodynamic quantities: the Debye temperature Θ relates to specific heat, dynamic properties, and melting temperature of solids, and the Grü neisen parameter γ describes the anharmonic effects in the vibrating lattice. The two quantities at various pressures and different temperatures are listed in Table 2. It can be seen clearly from Table 2 that, for a given temperature, Θ increases and γ decreases with increasing pressure. As the applied pressure varies from 0 to 50 GPa, Θ increases by 29.6%, 29.9%, 30.7%, 31.8%, 33%, 34.3%, and 35.8%, and γ decreases by 20%, 20.3%, 20.7%, 21.2%, 21.9%, 22.5%, and 23.3% at temperatures of 0, 200, 400, 600, 800, 1000, and 1200 K, respectively. For a given pressure, Θ decreases and γ increases with increasing temperature. As the applied temperature changes from 0 to 1200 K, Θ decreases by 7.1%, 5.4%, 4.3%, 3.6%, 3%, and 2.6%, and γ increases by 6.7%, 4.9%, 3.9%, 3.2%, 2.7%, and 2.4%, respectively. In addition, it is also found that, for a given temperature, Θ increases linearly and γ decreases almost exponentially especially for high temperature with increasing pressure. As pressure increases, the effects of temperature on them becomes less significant than pressure.
Table 2. Calculated Debye temperature Θ (in unit K) and Grü neisen parameter γ of 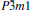 |
The temperature dependence of the calculated specific heat at constant volume CV and specific heat at constant pressure CP at different pressures are investigated, and the results are shown in Fig. 7. At low temperature, the difference of between CV and CP is very small, and the increases of CV and CP obey the harmonic approximations of the Debye model (∼ T3). However, the anharmonic effect on CV is suppressed at sufficient high temperature, and CV is close to a constant value which is called the Dulong– Petit limit (CV(T) ∼ 3R for monoatomic solids), and CP increases monotonously with the enhancement of the temperature. From Figs. 7(b) and 7(d), it is also found that CV and CP decrease with pressure at a given temperature and CV and CP increase with temperature at a given pressure, therefore, temperature has a more significant influence on the specific heat than pressure.
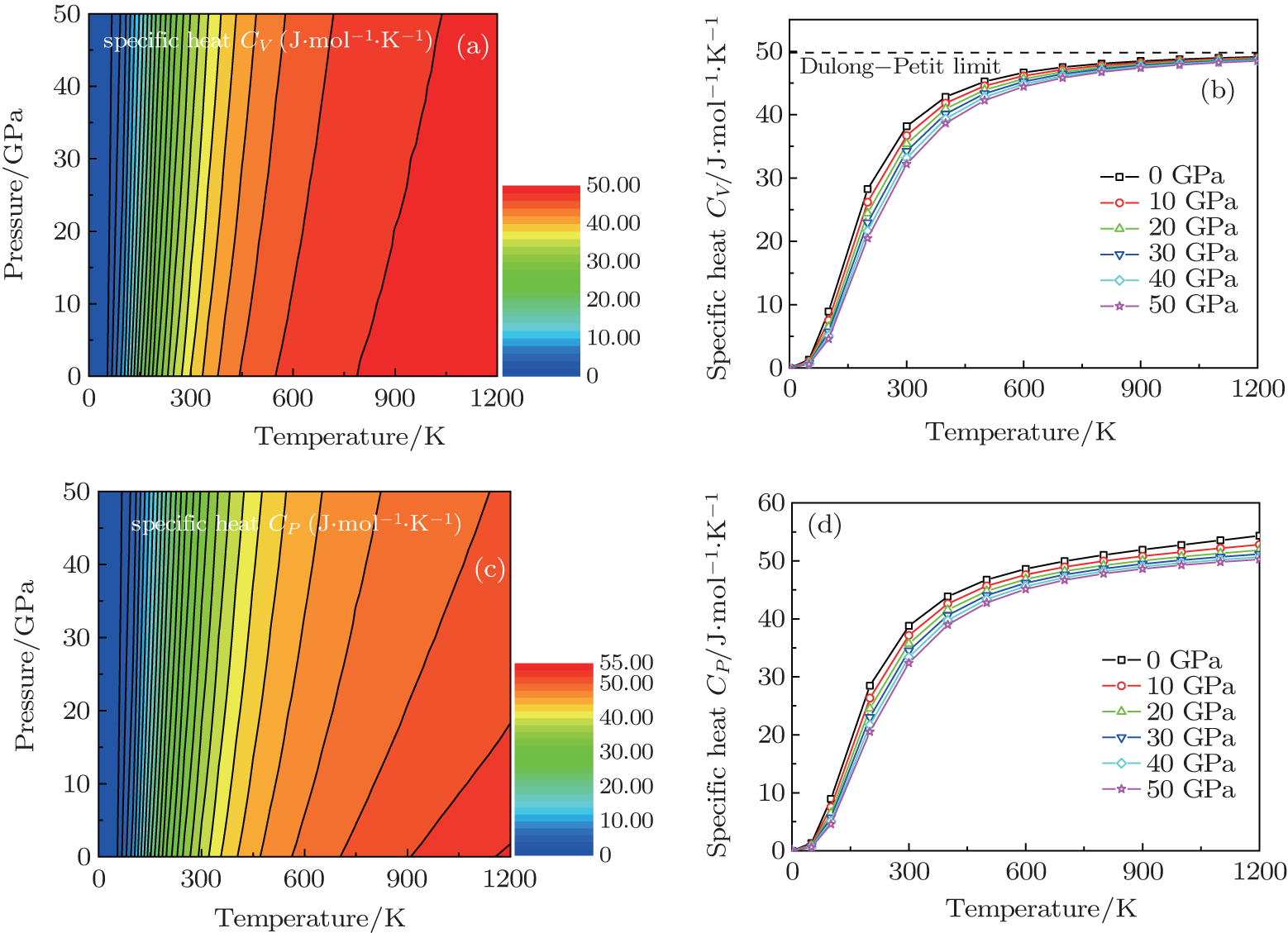 | Fig. 7. Temperature dependence of the calculated specific heat at constant volume CV and specific heat at constant pressure CP at different pressures for 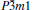 |
The temperature and pressure dependences of the thermal expansion coefficient α are displayed in Fig. 8(a)– 8(b), respectively. It can be seen from Fig. 8(a) that α increases steeply at low temperature especially for the case of 0 GPa, then gradually approaches to a linear increase at high temperature, finally becoming gentle at sufficiently high temperature. The influence of pressure on α is relatively small at low temperature, whereas the influence is enhanced obviously at high temperature. As pressure increases, α decreases rapidly at a given temperature and the effect of the temperature on it becomes less marked. From Fig. 8(b), one can also find that α tends to a constant value at high temperature and pressure. All of these results are similar to those of many kinds of materials by the Debye theory, such as ReB2, [50] OsB4, [51] PtAs2, [52] and FeB4.[53]
 | Fig. 8. (a) Temperature dependence of the thermal expansion coefficient α for  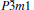 |
In conclusion, a hexagonal 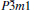

We acknowledge Prof. J J Zhao for his critical reading of the manuscript.
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|
| 35 |
|
| 36 |
|
| 37 |
|
| 38 |
|
| 39 |
|
| 40 |
|
| 41 |
|
| 42 |
|
| 43 |
|
| 44 |
|
| 45 |
|
| 46 |
|
| 47 |
|
| 48 |
|
| 49 |
|
| 50 |
|
| 51 |
|
| 52 |
|
| 53 |
|