
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Modeling the interaction of nitrate anions with ozone and atmospheric moisture
[Galashev A. Y.† ]
]
]
|
|


†Corresponding author. E-mail: alexander-galashev@yandex.ru
The molecular dynamics method is used to investigate the interaction between one–six nitrate anions and water clusters absorbing six ozone molecules. The infrared (IR) absorption and reflection spectra are reshaped significantly, and new peaks appear at Raman spectra due to the addition of ozone and nitrate anions to the disperse water system. After ozone and nitrate anions are captured, the average (in frequency) IR reflection coefficient of the water disperse system increased drastically and the absorption coefficient fell.
In the Earth’ s atmosphere, the ozone layer is situated at an altitude of 20 km– 30 km. The ozone layer protects life on Earth from being directly exposed to hard ultraviolet (UV) radiation. Since ozone is a greenhouse gas, its content in the troposphere affects the global climate. Oxidation of trace components is often treated as a self-purifying ability of the atmosphere. Although there is no definition of oxidizing ability, it is often associated with an excess of OH. However, many other oxidants, including O2 and O3, and free radicals different from OH, can contribute to the oxidation process taking place in the atmosphere. The 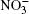
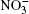
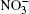
Nitrate ions are some of the most widespread ions in the Earth’ s atmosphere. Their proportion exceeds that of atmospheric Cl– ions by more than 3.5 times and, by hundreds of times, that of Br– ions.[1, 2] Many atmospheric processes are explained by their presence. In Ref. [3], solvation of nitrate ions was studied at the air– water interface. A nitrate ion was shown to prefer surface solvation to bulk solvation. The lifetime of 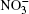

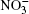
Nitrate ions are generated in Earth’ s atmosphere in a chain of chemical reactions with nitric oxide. The nitrate ions are captured by aerosols, fall down with precipitation and become fixed in polar ice. Experimental investigation of the air-aqueous nitrate interface cannot completely provide an insight into the role of nitrate ions at the surface of aqueous-phase atmospheric aerosols. However, computer simulation to a certain extent gives such an understanding. Absorptions of ozone, oxygen, methane, and chlorine and bromine ions by atmospheric moisture are investigated in the molecular dynamics model.[4– 12] There are no data on how the presence of 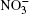
The aim of the present study is to investigate the simultaneous interaction of water clusters with nitrate ions and ozone molecules, and to reveal the behaviors of changes in spectral characteristics of disperse water systems, which result from such interactions.
Properties of clusters and interfaces are not critically evaluated by using the original TIP4P potential model of water.[13] For this reason a new potential including an optimal set of parameters that describe the important properties of aqueous clusters (binding energies and minimum energy structures) has been developed.[14] It turned out that structure and thermodynamic properties of the bulk and liquid/vapor interface of water are also well reproduced by using a modified potential. A modification of the model fulfilled in Ref. [14] was connected with a considerable change in parameters of the Lennard-Jones (LJ) part of the potential; as a consequence, the coefficients with the terms describing repulsion and attraction were reduced by factors 2.5 and 2.9, respectively. Additionally, in this model, a negative charge was located at a point 0.0215 nm away from the oxygen nucleus instead of 0.015 nm as previously. This displacement allowed the permanent dipole moment of the water molecule to be corrected to 1.848 D, which corresponds to the experimental value in a gaseous phase. In the present work, a polarizable variant of the improved TIP4P model of water is employed.
Interactions between nitrate ions and between water and 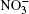


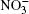
A pair of used atom– atomic potentials was treated as Lennard-Jones and Coulomb contributions. The parameters of the potential for the description of these interactions are given in Refs. [15] and [17]. In molecular dynamics calculations using the Stillinger– David polarization model, [19] the potential energy U of a cluster (H2O)50 took the value − 24.25 eV at T = 40 K.[20] In the model presented herein, the U value for the (H2O)50 aggregate is − 23.1 eV at T = 237 K.
The interaction of a water cluster with ozone molecules and nitrate ions was studied at the same temperature 237 K. Initially, the center of a free ozone molecule was located at a distance of 0.6 nm– 0.7 nm from the nearest center of the water molecule incorporated in the cluster. As this took place, the O3 molecule had a spontaneous orientation. Ions were brought to the system 6O3 + (H2O)50 in pairs and arranged on coordinate axes on different sides of the system at a distance of not less than 0.6 nm from any atom of the nearest molecule. The cutoff radius of molecular interactions in the model was 0.9 nm.
In the present work, we use a canonical ensemble simulation to calculate the spectral characteristics of water clusters.[21, 22] The molecular dynamics calculation is carried out in time steps of Δ t = 0.2 × 10− 16 s. The calculation of spectral characteristics is begun after the system has achieved an equilibrium state, which takes a time of tne = 3 × 105Δ t (6 ps). The (O3)6(H2O)50 cluster is also formed in ∼ 3 × 105Δ t time steps regardless of the presence of 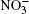
Flexible models of molecules are considered. Molecular flexibility is achieved by using the procedure designed within the framework of the Hamiltonian dynamics in Refs. [23]– [25]. In the model, each molecule can be considered as a polarizable point dipole situated in the center of mass of the molecule.[4– 12] The Gear fourth-order method is employed to integrate the equations of motion of centers of mass of molecules.[26] The analytical solution of motion equations for molecular rotation is accomplished by using the Rodrigues– Hamilton parameters; [27] and the integration scheme of motion equations considering rotations corresponds to the approach proposed by Sonnenschein.[28]
The absorption cross section of IR radiation is defined as[29]

where n is the frequency-independent refractive index, ε v is the vacuum dielectric permittivity, and c is the velocity of light.
In Eq. (1), the sum of vector differences of individual molecular dipoles is defined as

In the case of unpolarized light, the Raman spectra is given by the relation[29]
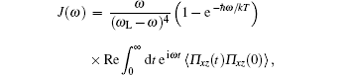
where
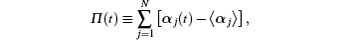
ω L is the frequency of an exciting laser, Π xz represents the xz component of the Π (t) value, the x axis is directed along the molecular dipole, and xy is the molecular plane.
The reflection coefficient R is defined as the ratio between the average energy flux reflected from the surface and the incident flux. At normal incidence of a plane monochromatic wave, the reflection coefficient is described by the formula[30]

It is supposed here that the incidence of a wave occurs from the transparent medium (medium 1) into a medium that can be either transparent or not, i.e., absorbing and scattering medium (medium 2). The indices of the dielectric permittivity in Eq. (2) refer to those of the medium.
We denote the systems as I (H2O)n, n = 10, 15, … , 50; 
The configurations of 
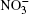
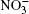
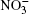
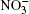

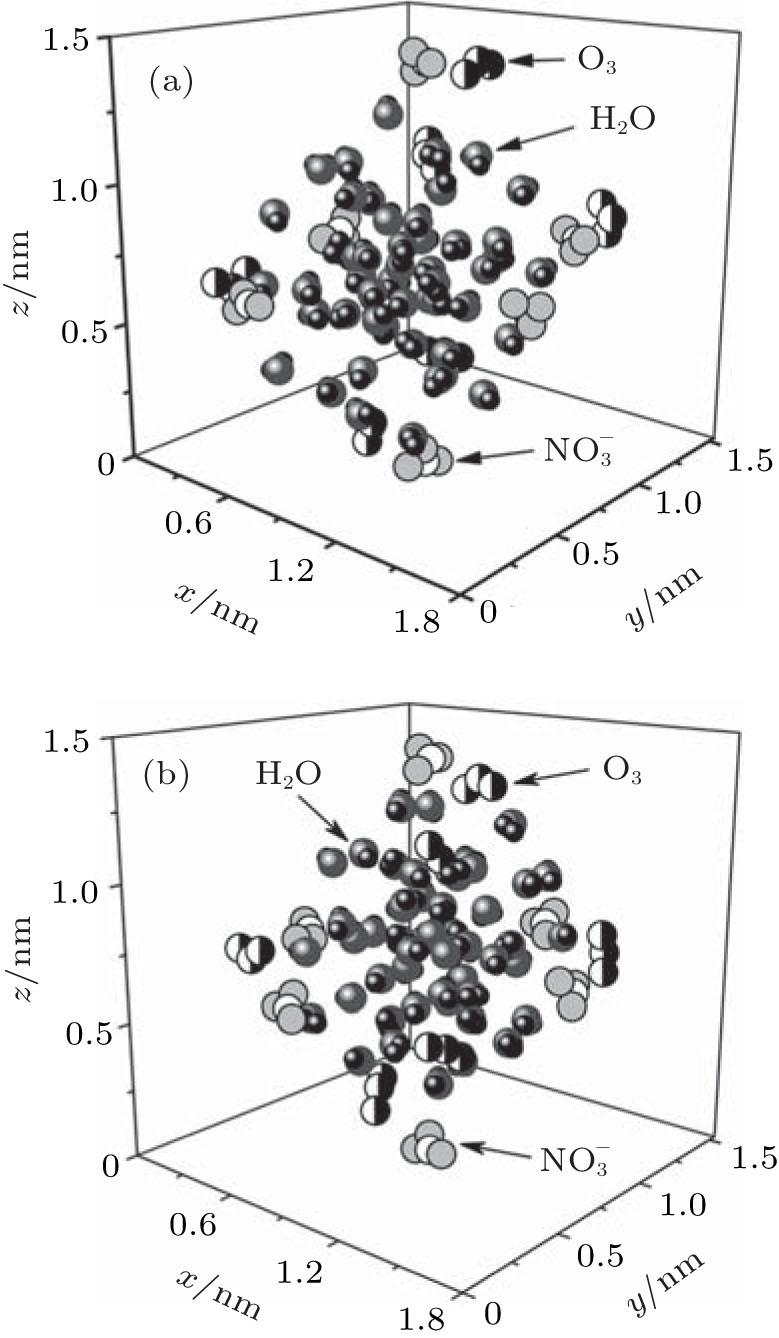 | Fig. 1. Configurations of the cluster  |
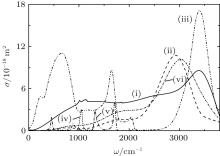
Figure 2 displays IR absorption spectra for systems I and II, and it also shows the experimental IR spectra of liquid water[31] and spectra of O3[32] and HNO3, [33] which are present in the troposphere. The positions of the most intense bands in the IR spectrum for the water cluster system (system I) and in the spectrum of liquid water (3389 cm− 1 and 3404 cm− 1) correlate well with each other. The presence of 
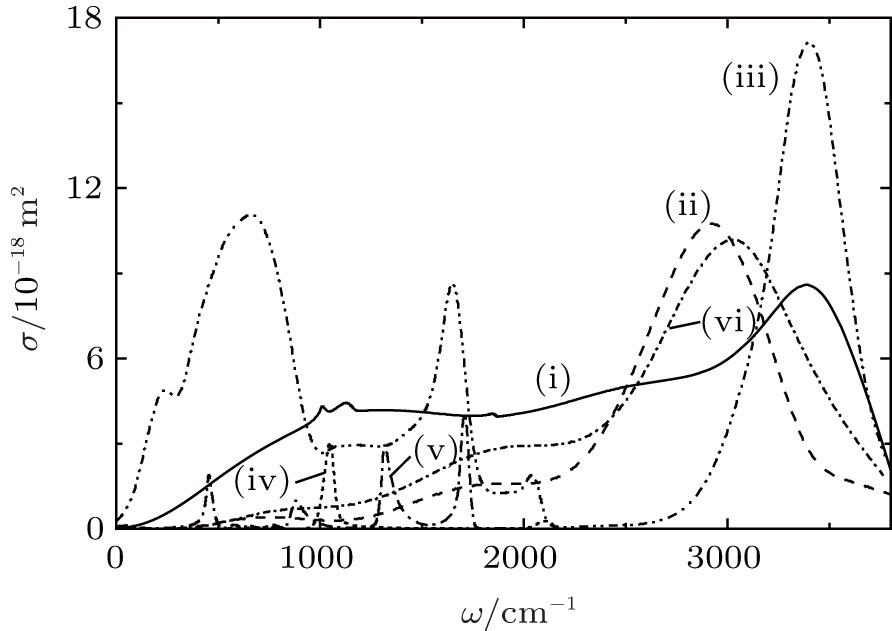 | Fig. 2. IR absorption spectra of the systems (i) I, (ii) II (for (i) and (ii) T = 237 K); (iii) liquid water (T = 293 K), the experiment; [31] (iv), (v) stratospheric measurements of O3[32] and HNO3, [33] respectively; (vi) system II at T = 287 K. |
The most intense band in the IR spectrum of liquid water appears due to the superposition of three modes: ν 1 is the symmetric stretching vibration, ν 3 is the antisymmetric stretching vibration, and the overtone of a mode ν 2 is the deformation vibration (bending of covalent bonds). The mode ν 2 is responsible for the emergence of a band at 1644 cm− 1. The band at 690 cm− 1 is formed due to librations determined by restrictions imposed by hydrogen bonding. The weak band at 200 cm− 1 is associated with translation vibrations, including the stretching of the O– H … O hydrogen bond and its bending. At the IR spectrum of the water cluster system I, a blue shift of the libration mode by ∼ 380 cm− 1 with respect to the corresponding band in the IR spectrum of bulk water is observed. This band of system I is positioned between the positions of the second and the third peaks in the IR spectrum of stratospheric HNO3.
The IR spectrum of system II at T = 237 K shows three broad bands. The first one, of weak intensity, is localized at 748 cm− 1, the second, more intensive, band is at 1842 cm− 1, and the third one, most intensive, is located around 2936 cm− 1. The position of the first band agrees with quantum-mechanical calculation of localization of the mode of deformation vibrations (720 cm− 1) in the 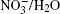
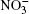
The work[36] showed that the absorption by (H2O)50 clusters of both molecular and monatomic oxygen yields a reduction in the integral intensity Itot of the IR absorption spectrum by a factor of 1.5– 2. In the course of the study, IR absorption spectra were calculated for the disperse water system III with nitrate ions 
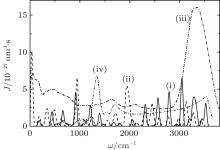
The Raman spectra of systems I and II are represented by a great number of well-resolved peaks (Fig. 3). The three most intense peaks of system II fall at the frequencies of 938, 1941, and 3125 cm− 1. The most intense band in the Raman spectrum of disperse water system I is localized at 3040 cm− 1. The Raman spectrum of liquid water[37] shows the most intense band at 3337 cm− 1, and the Raman spectrum of complexes 
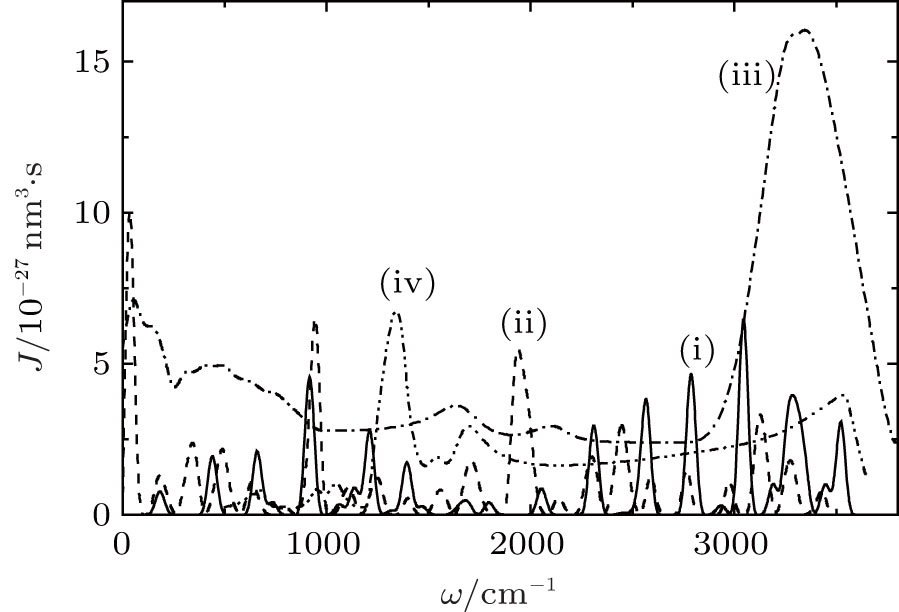 | Fig. 3. Raman spectra for the systems (i) I, (ii) II (for (i) and (ii) T = 237 K); (iii) liquid water (T = 293 K), the experiment; [37] (iv) Raman spectrum of the complexes  |
Characteristic vibrations in liquid water occur with average intensities of 200 cm− 1 and 686 cm− 1. With allowance for anharmonicity of vibrations, one can expect that the sum of these modes will result in the occurrence of a band at the Raman spectrum localized around a frequency of 900 cm− 1. The intensity of the band at 938 cm− 1 in the Raman spectrum of system II increases with respect to the corresponding characteristic in the spectrum of system I (a band at 917 cm− 1). This increase occurs due to the influence of 
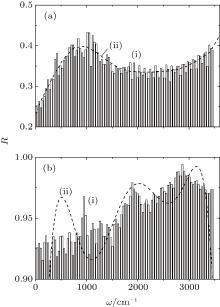
Figure 4 shows the frequency spectra of the reflection coefficient of disperse water systems I and II. The maximum of the R-spectrum of the system (I) of pure water clusters falls at a frequency of 1041 cm− 1, and that of the spectrum of the system (II) adsorbing ozone and nitrate ions localizes at 2882 cm− 1. The disperse water system I has an average (in frequency) IR reflection coefficient of R̄ = 0.35, while the same parameter for system II is 0.94. The spectrum R(ω ) for system I is fitted by a curve with a single pronounced peak, and the corresponding spectrum for system II is approximated by a frequency dependence with three peaks. Thus, homogeneous deposition of ozone molecules and nitrate ions onto the surface of (H2O)50 clusters produces high-efficiency reflection of IR radiation by the cluster system.
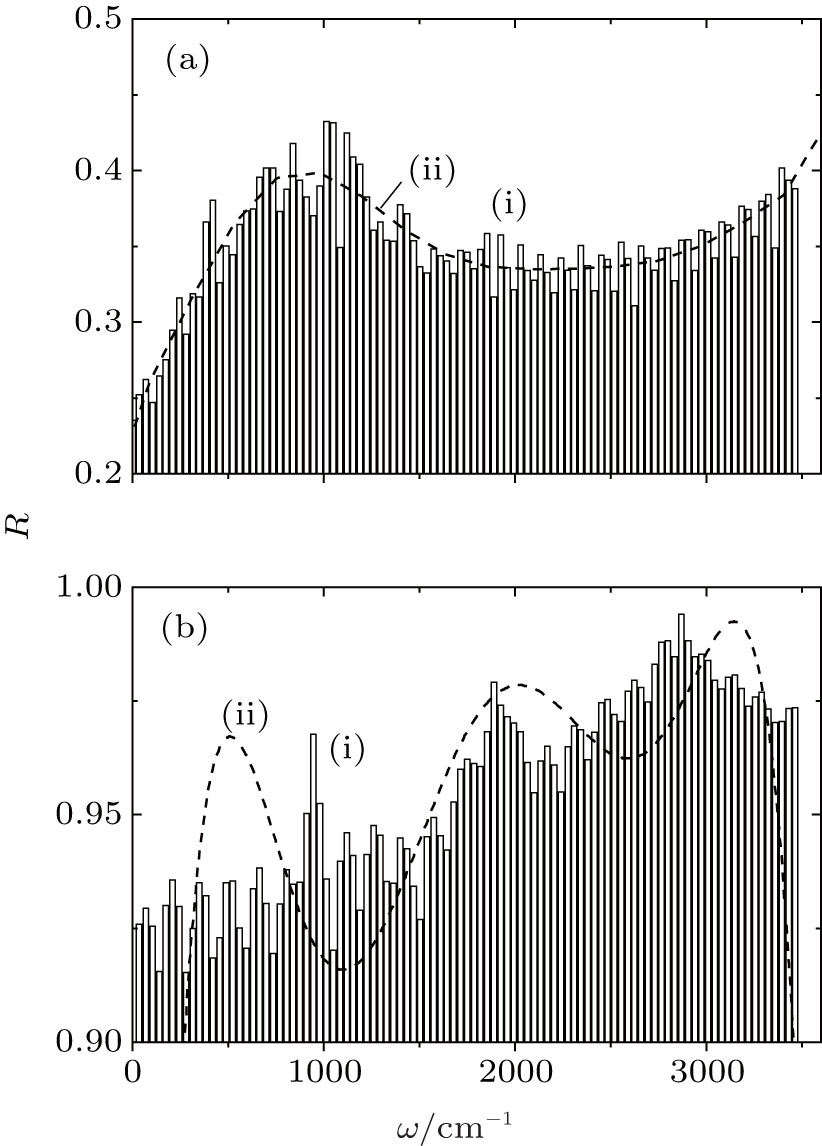 | Fig. 4. Reflection coefficients of IR radiation for the cluster systems at T = 237 K: (a) system I, (b) system II; (i) MD calculations, (ii) approximation of the calculated dependence by the polynomial R(ω ) of the sixth order. |
In the present work, the mechanism of adsorption of nitrate ions and ozone by water clusters is investigated. The initial orientation of surface water molecules in a cluster with hydrogen directed outwards produced an attractive force for 
Ozone is a very powerful oxidizer, much more powerful than oxygen. Therefore, the following processes should actually take place further at the surface of a cluster that has adsorbed ozone and ions. At the high concentrations observed, ozone decomposes into ordinary diatomic and monatomic oxygen. Under atmospheric conditions its half-life is approximately 30 min. Monatomic oxygen forms NO2 and O2 reacting with 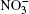
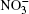
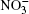
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|
| 35 |
|
| 36 |
|
| 37 |
|
| 38 |
|
| 39 |
|